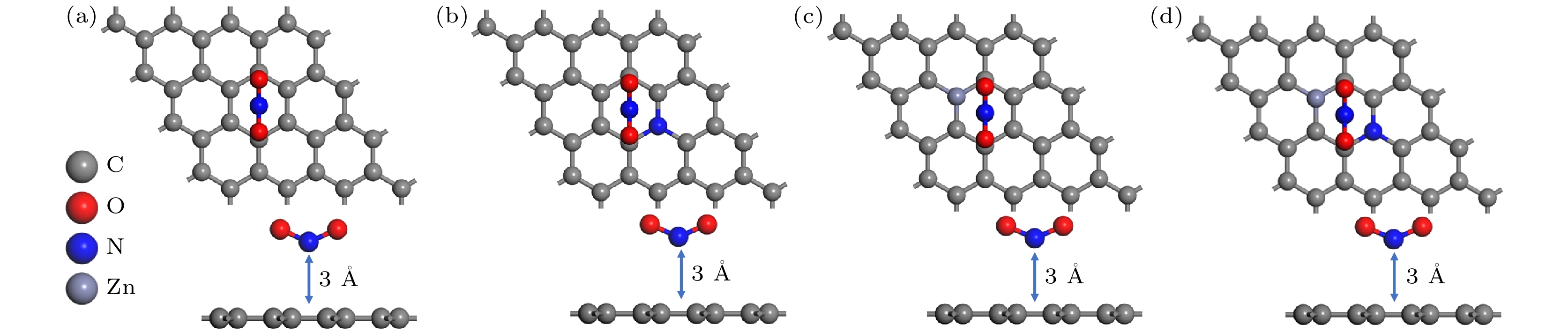
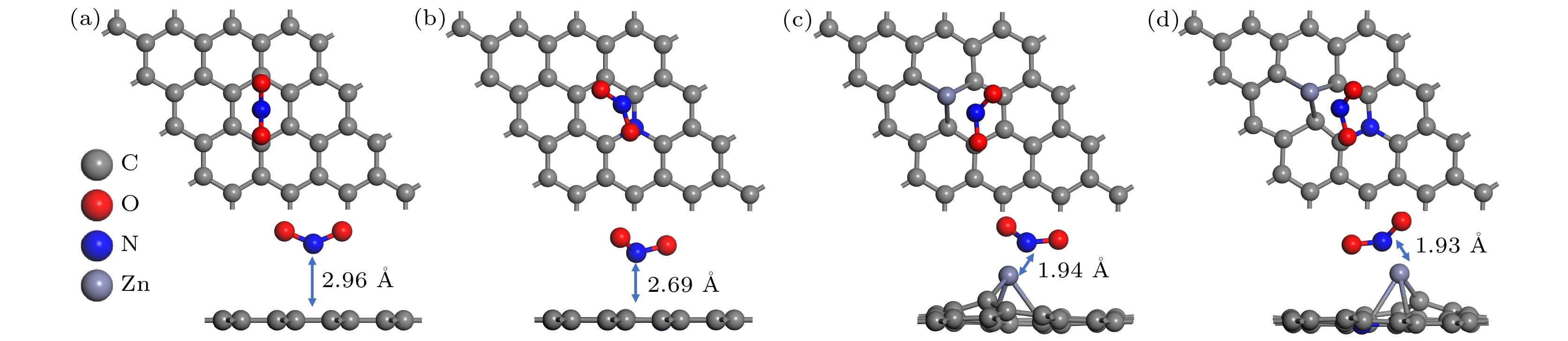
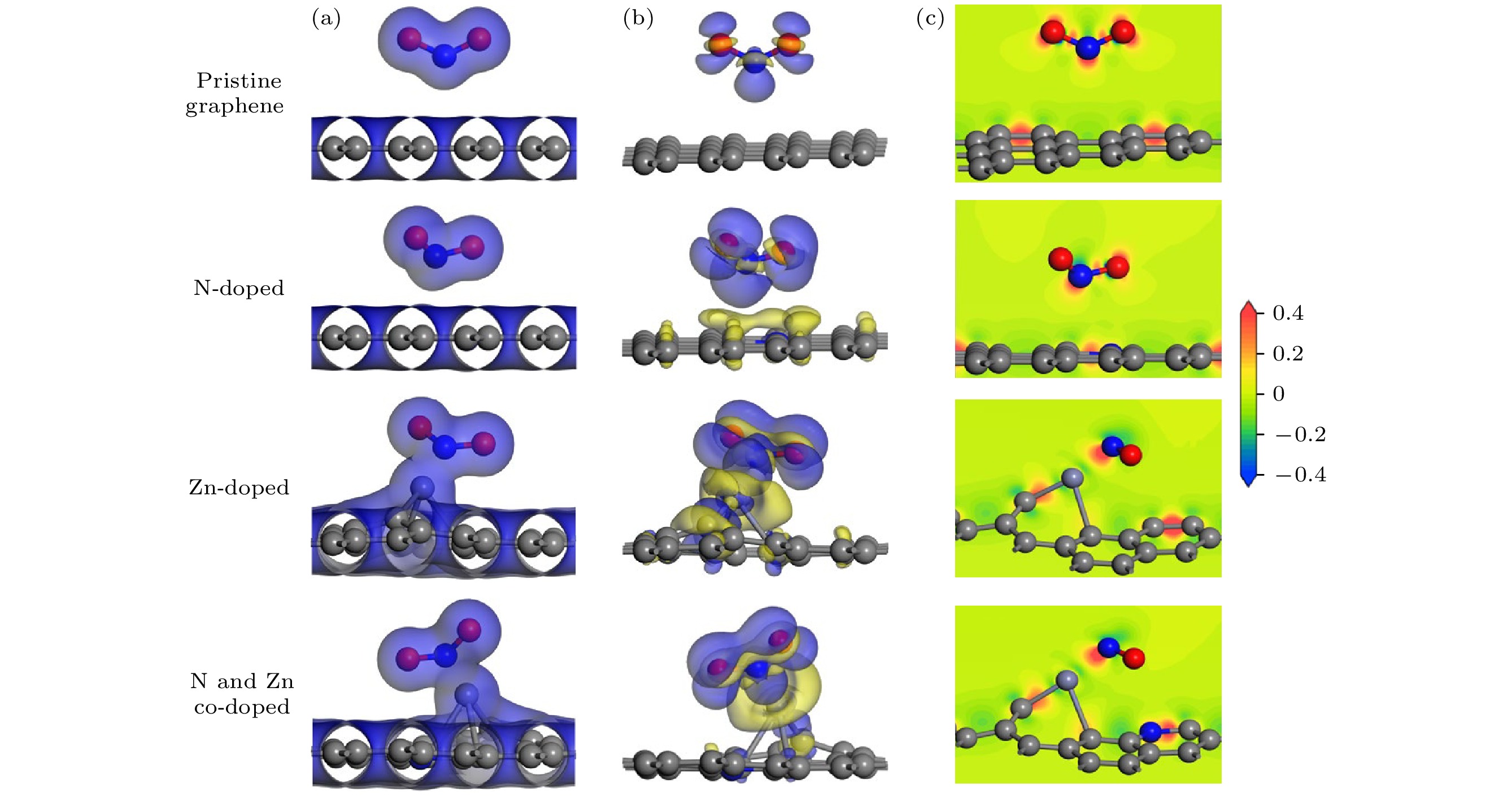
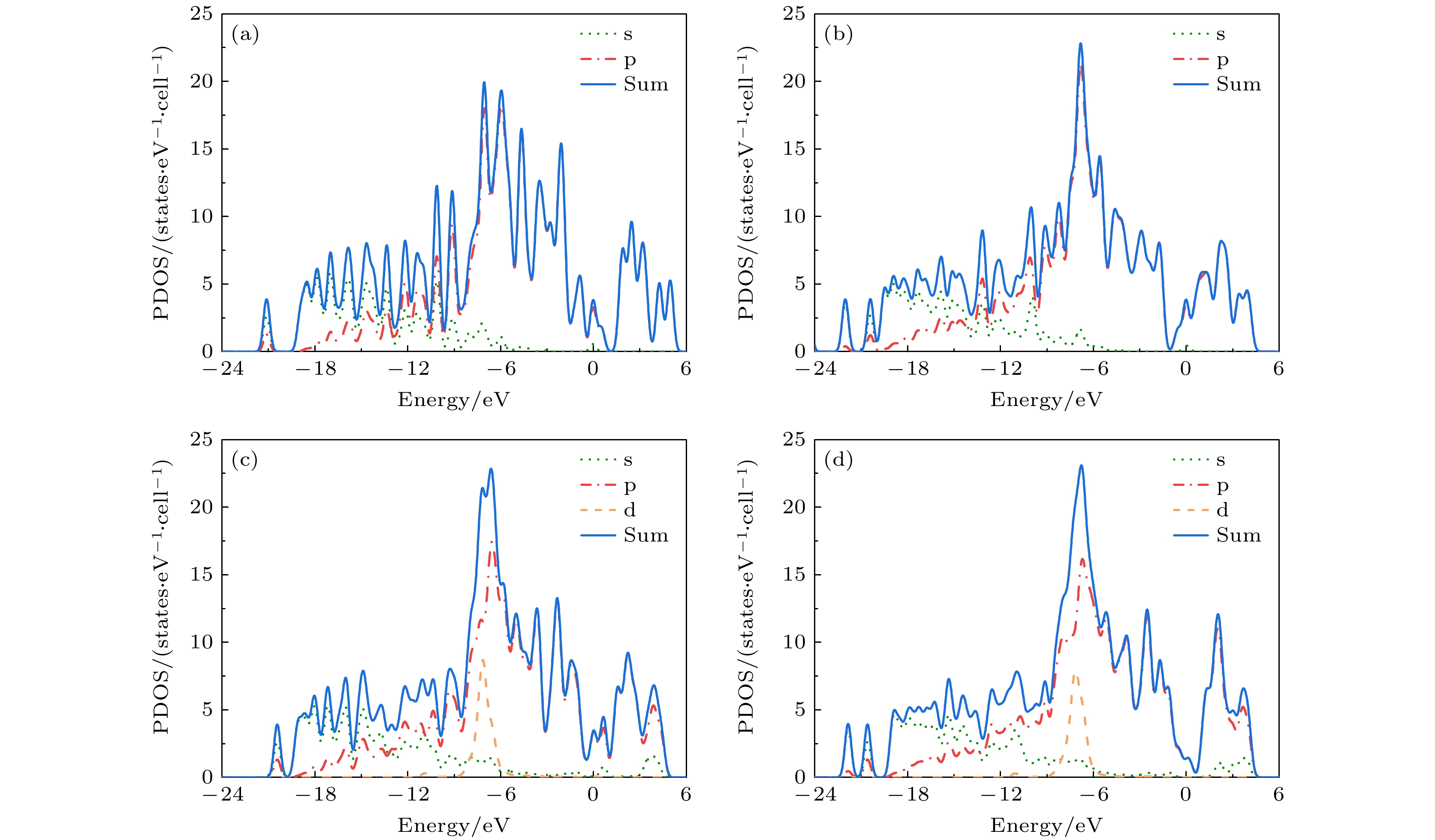
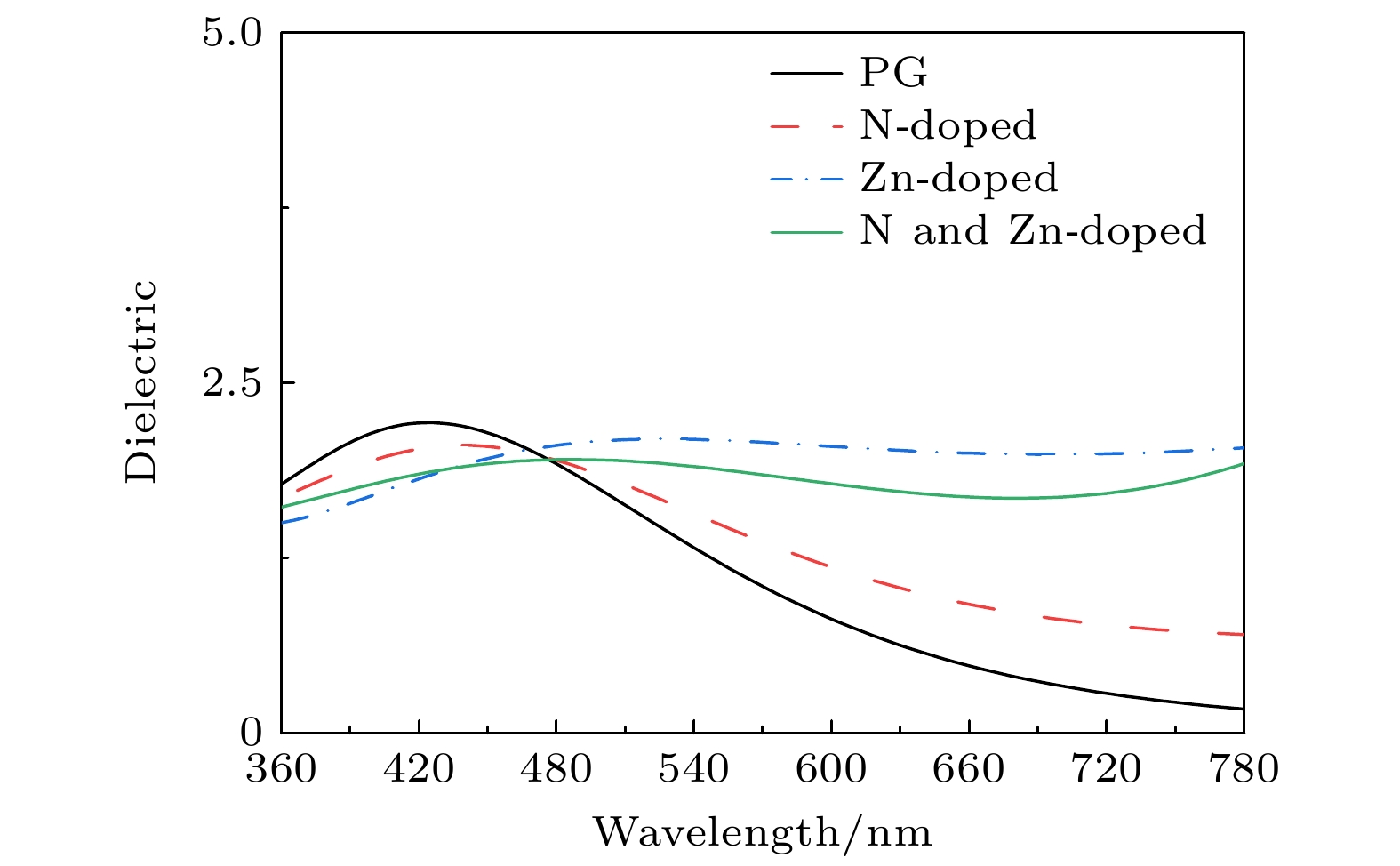
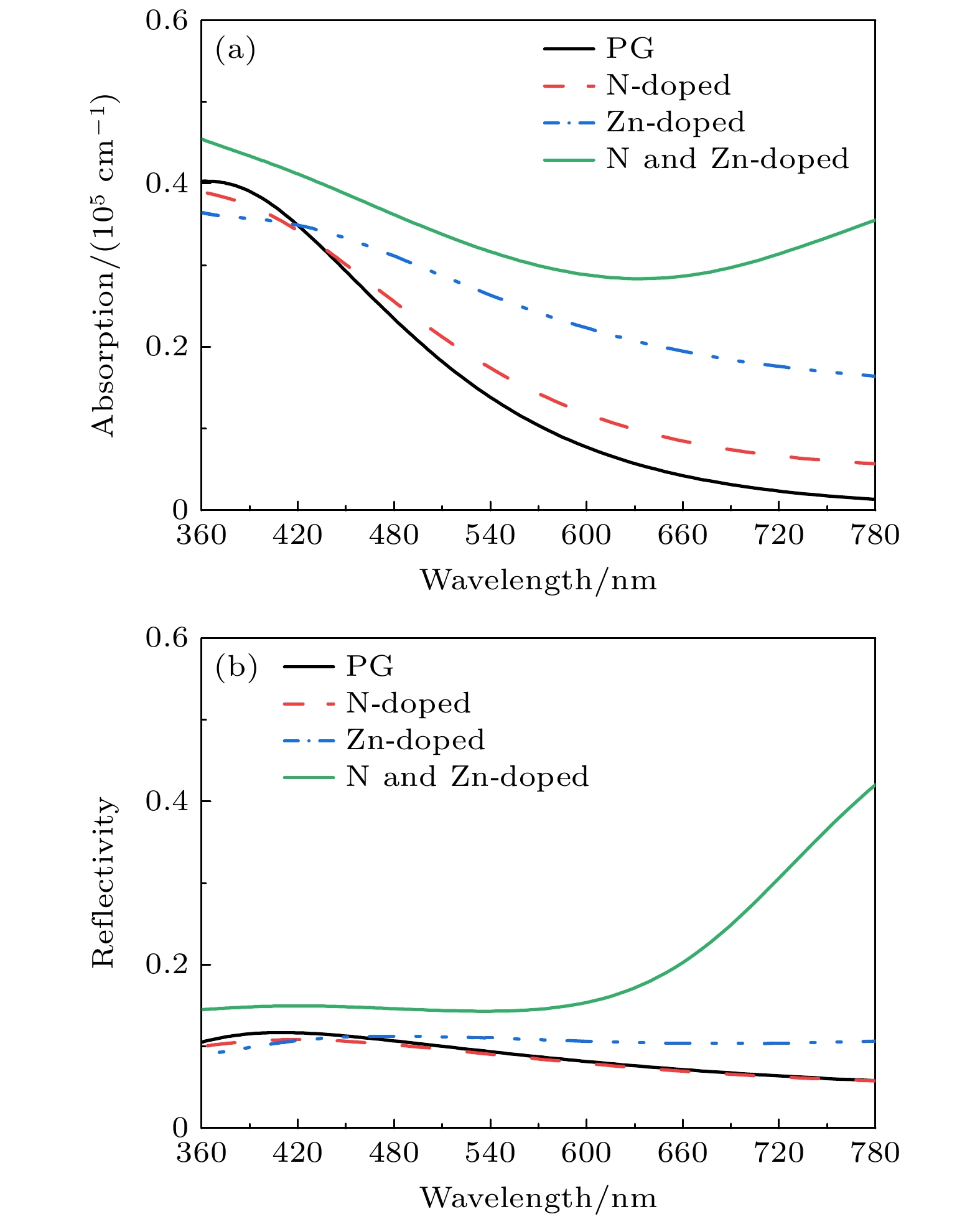
In order to study the adsorption of NO2 on pristine graphene and doped graphene (N-doped, Zn-doped, and N-Zn co-doped), we simulate the adsorption process by applying the first-principles plane-wave ultrasoft pseudopotentials of the density-functional theory in this work. The adsorption energy, Mulliken distribution, differential charge density, density of states, and optical properties of NO2 molecules adsorbed on the graphene surface are calculated. The results show that the doped graphene surface exhibits higher sensitivity to the adsorption of NO2 compared with the pristine graphene surface, and the order of adsorption energy is as follows: N-Zn co-doped surface > Zn-doped surface > N-doped surface > pristine surface. Pristine graphene surface and N-doped graphene surface have weak interactions with and physical adsorption of NO2. Zn-doped graphene surfac and N-Zn co-doped graphene surface form chemical bonds with NO2 and are chemisorbed. In the visible range, among the three doping modes, the N-Zn co-doped surface is the most effective for improving the optical properties of graphene, with the peak absorption and reflection coefficients improved by about 1.12 and 3.42 times, respectively, compared with pristine graphene. The N-Zn co-doped graphene not only enhances the interaction between the surface and NO2, but also improves the optical properties of the material, which provides theoretical support and experimental guidance for NO2 gas detection and sensing based on graphene substrate.












 DownLoad: CSV
DownLoad: CSV


 DownLoad:
DownLoad:




