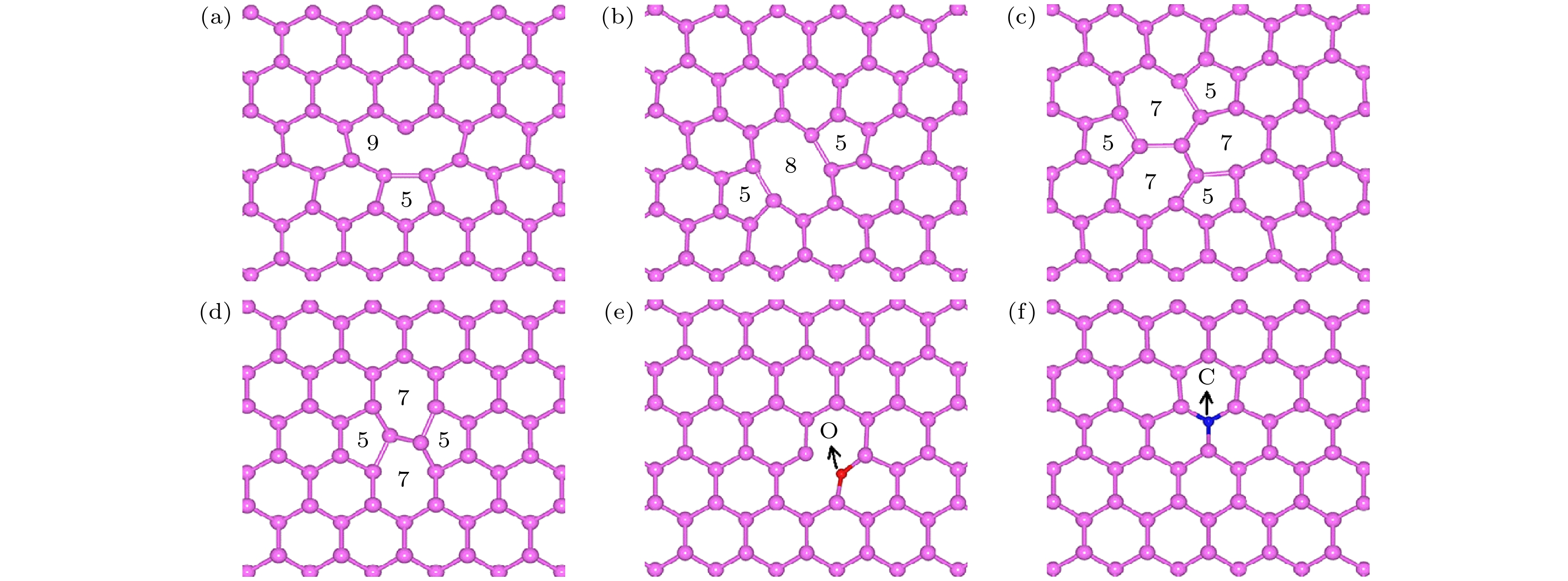
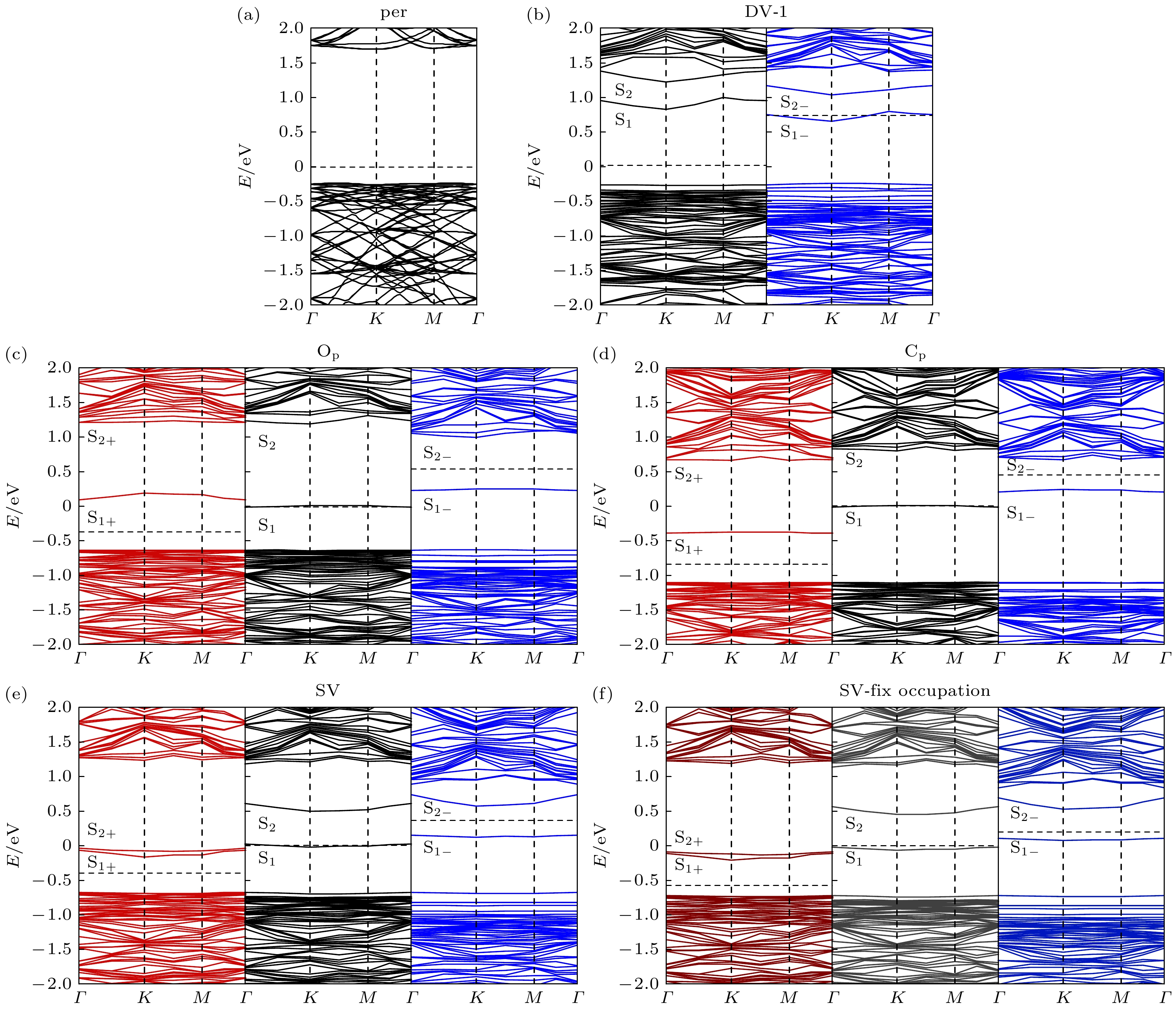
As a new two-dimensional material, blue phosphorus has attracted considerable research interest due to its high carrier mobility and large bandgap. Although the structural defects of blue phosphorus have been discussed recently, the charged properties of these defects have not been explored. In this paper, using first-principles calculations based on density functional theory, the six most stable point defects and their corresponding charged states in blue phosphorus are studied, including Stone Wales (SW), single vacancy (SV), two double-vacancy (DV-1 and DV-2) and two substitution defects (O
Pand C
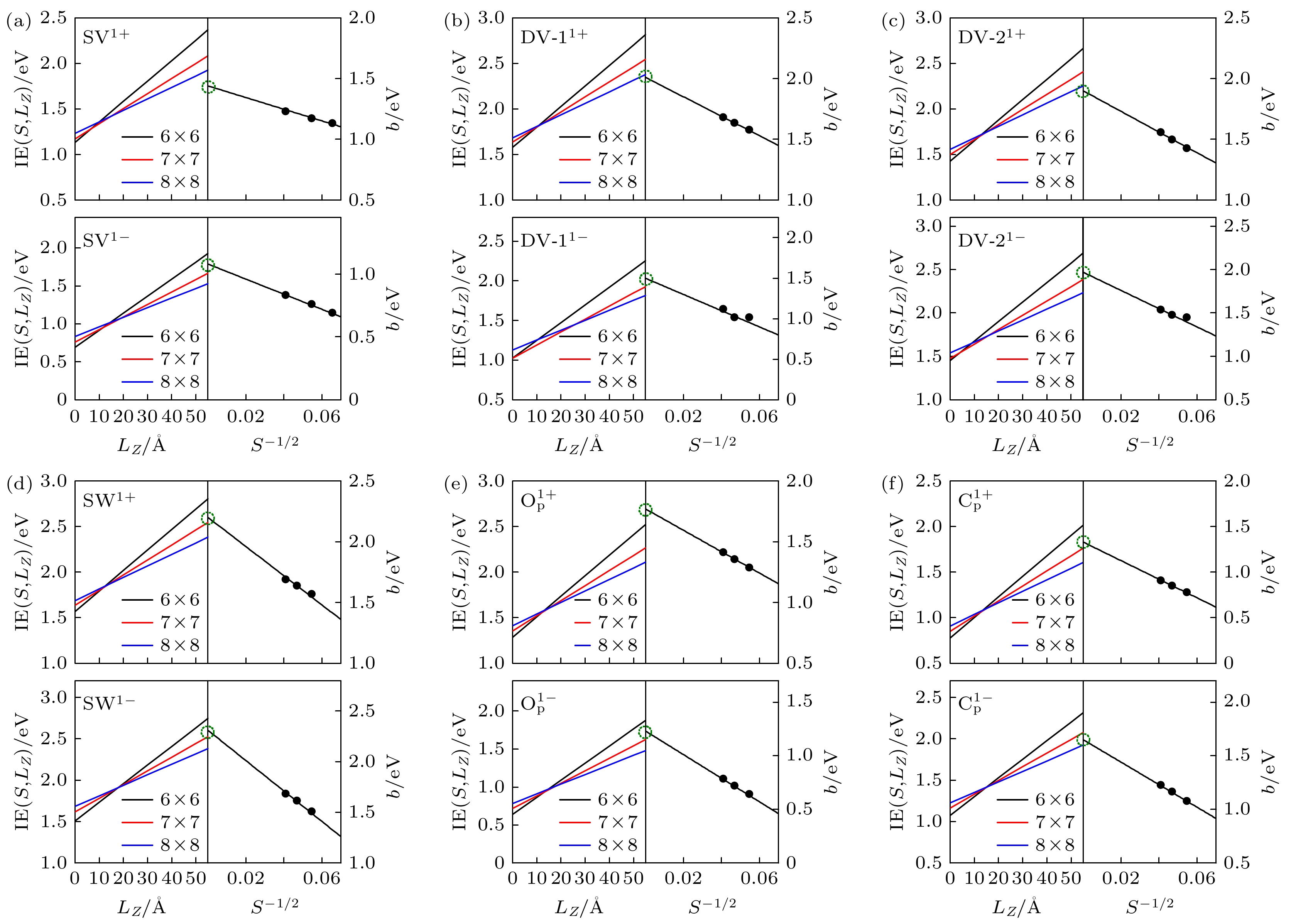
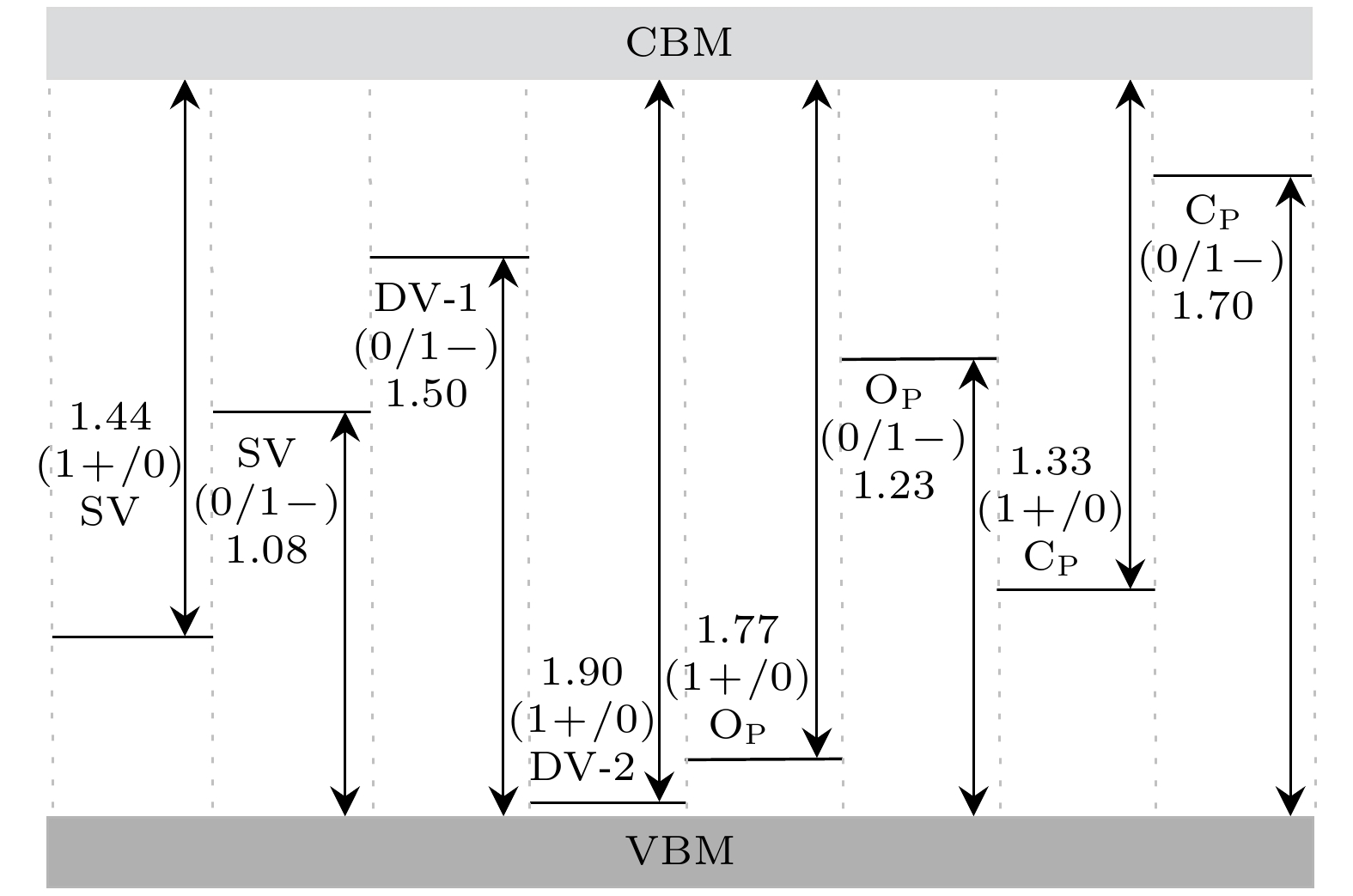
P). The converged ionization energy values of charged defects in blue phosphorus are obtained by extrapolating the asymptotic expression of the energy dependent on the cell size. Subsequently, the formation energy values for different charge states are modified to determine their structural stabilities. Finally, their electronic properties are analyzed through band structures. The results suggest that SV
1–is easy to ionize, owing to its lowest ionization energy (1.08 eV). Furthermore, among the defects we are considering, O
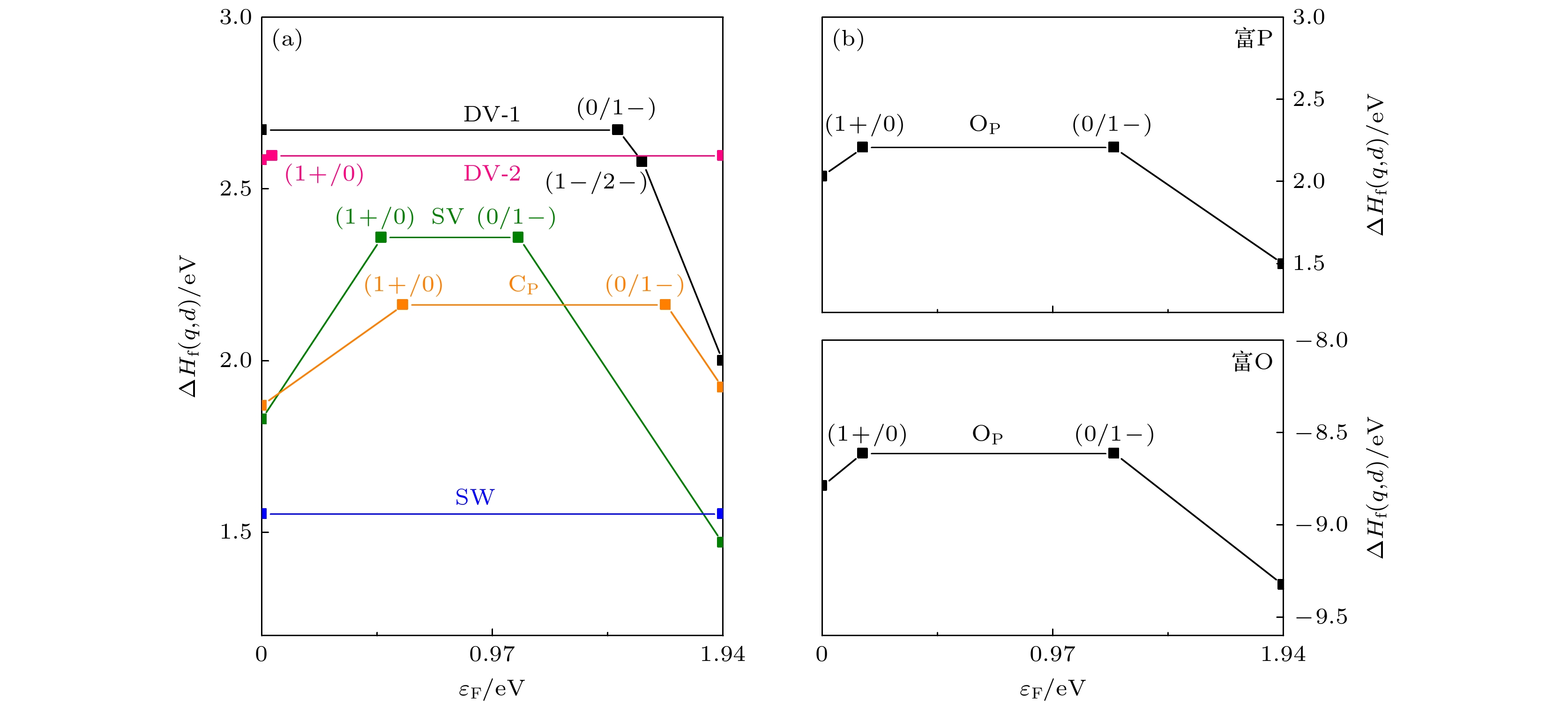
P
1–is the most stable charged defect in blue phosphorus, with the lowest formation energy (–9.33 eV) under O-rich chemical potential condition. The negative formation energy indicates that O atoms can exist stably in blue phosphorus, implying that blue phosphorus is easily oxidized. The introduction of defect states will affect the bandgap of blue phosphorus, and the ionization of defects will cause the defect energy levels to shift, leading defects to transition between shallow and deep levels. This study provides theoretical guidance for the application of defect engineering in two-dimensional materials.














 DownLoad:
DownLoad:



