In recent decades, significant progress has been made in the precise theoretical investigation of gas-phase chemical reactions. Presently, a major challenge in the field of quantum dynamics is to develop the precise methodologies for studying chemical reactions involving more than four atoms. As a typical multi-atomic reaction system, the F+CH
4reaction and its isotopic substitution reactions have attracted widespread attention from both experimental and theoretical perspectives in recent years. Experimental studies on the reaction of F+CHD
3have revealed that the stretching vibration excitation of the C—H bond inhibits the bond dissociation, favoring the formation of DF+CHD
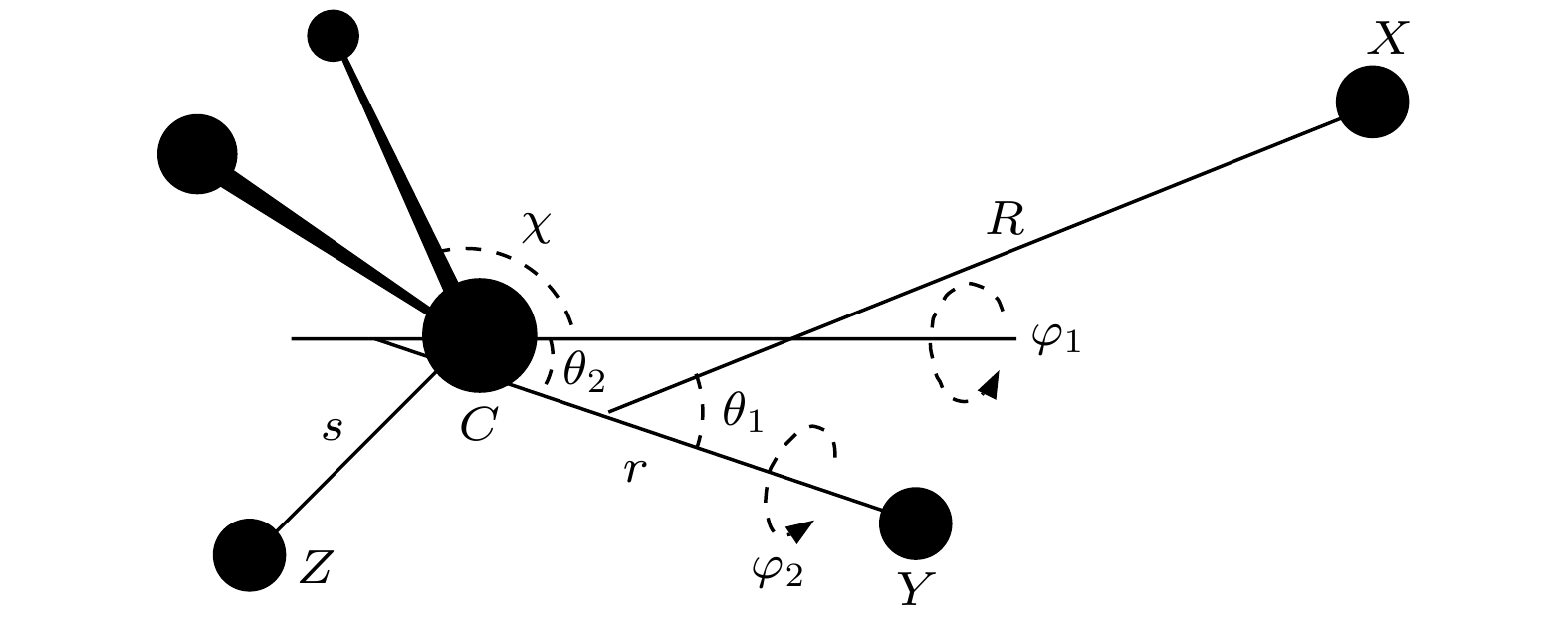
2product channels. In this study, we use a seven-dimensional quantum time-dependent wave packet method to investigate the dynamics of the F+CHD
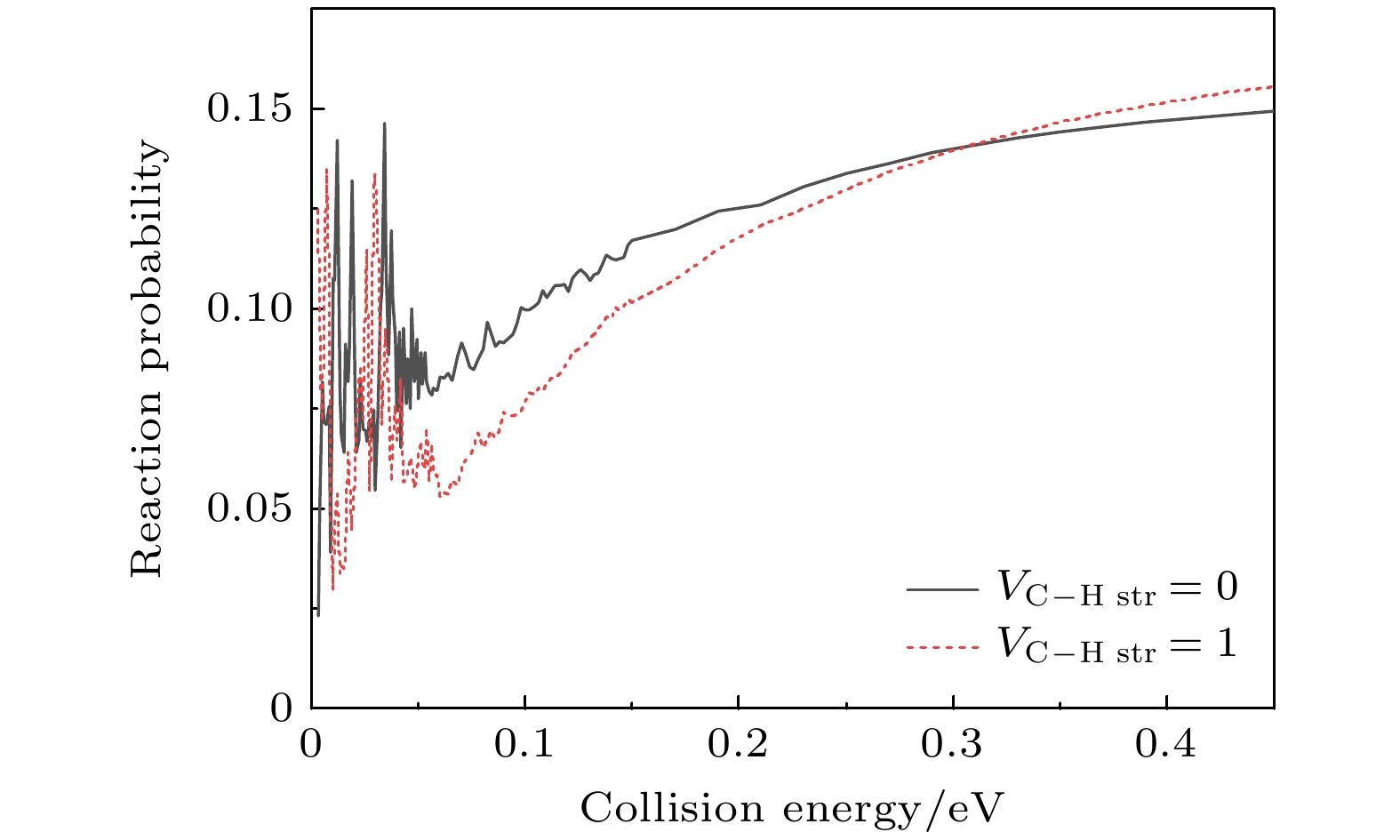
3reaction in both the reactant vibrational ground state and the first stretching excited state of the C—H bond. In this work, the reaction probabilities under different vibrational conditions are analyzed, showing that when the collision energy is below 0.06 eV, the reaction probability curves exhibit numerous fast-oscillating peaks, supporting the experimentally suggested phenomena of dynamic resonance. In a collision energy range from 0.06 eV to 0.3 eV, the reaction probability for the HF product channel in the vibrational excited state is lower than that in the ground state, which is consistent with experimental observation. Through the analysis of the time-independent wave functions of product channels under low-energy collision conditions, it is found that for reactions involving vibrational ground states, the HF products in the product asymptotic region and the reaction transition state region are in the
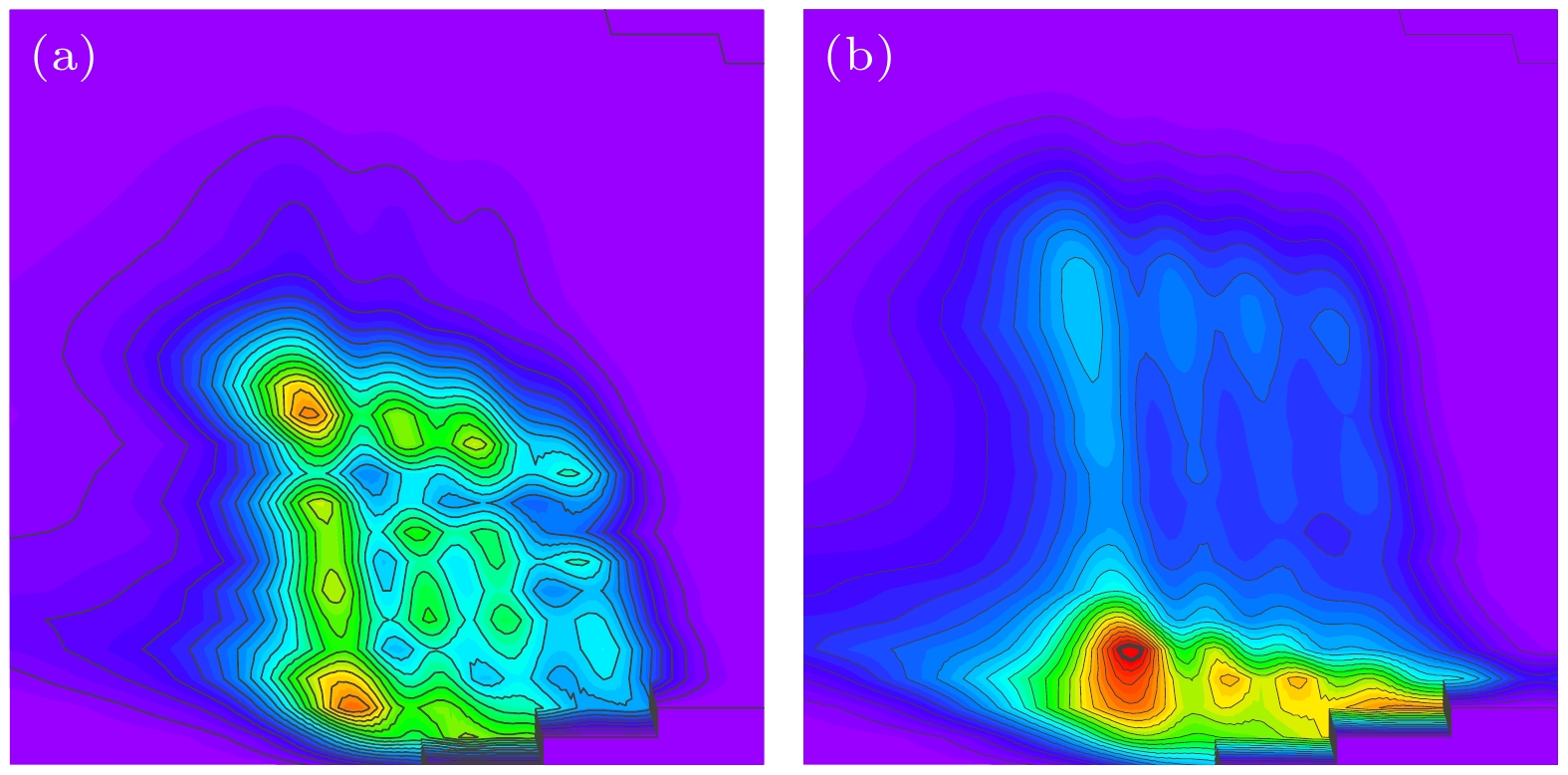
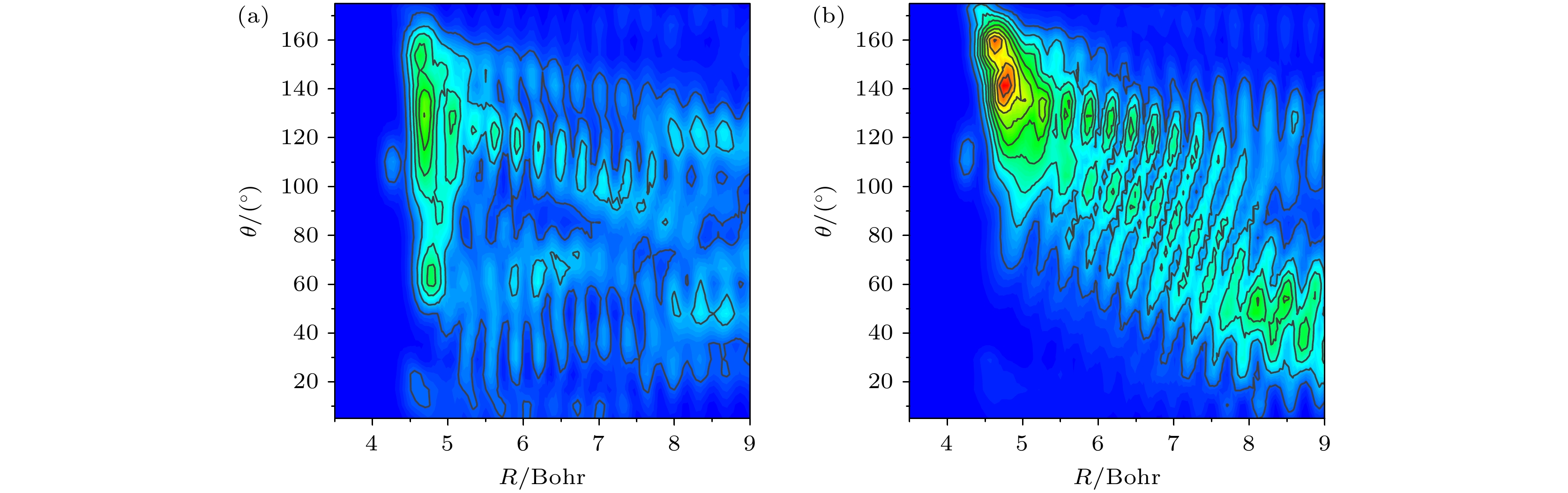
v'= 2 excited state and
v'= 3 excited state of stretching vibration, respectively, which are consistent with previous experimental observations and six-dimensional quantum wave packet simulations. For reactions involving the first excited state of C—H stretching vibration, the HF products in the product asymptotic region and the reaction transition state region are both in the
v'= 3 excited state of stretching vibration, which are consistent with the results obtained based on energy analysis. Simulation results indicate that in the case of low-energy collisions, the time-independent wave function for the C—H stretching vibrational excited state tends to be closer to the D atom side in the transition state region. This phenomenon is attributed to the more significant energy advantage of the vibrational excited state potential energy surface in the large collision angle region, explaining the inhibitory effect of stretching vibration excitation on the HF product channel. This study offers important theoretical support for explaining experimental results and contributes to a more in-depth understanding of the influence of vibrational mode excitations on the dynamical processes in poly-atomic reactions.












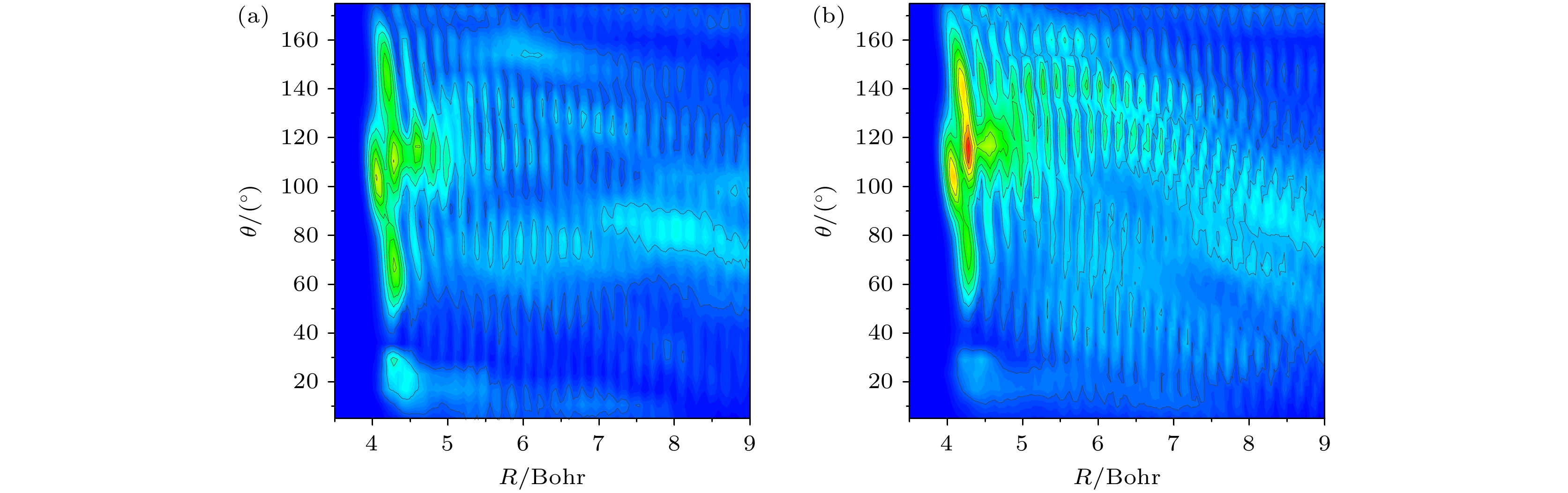
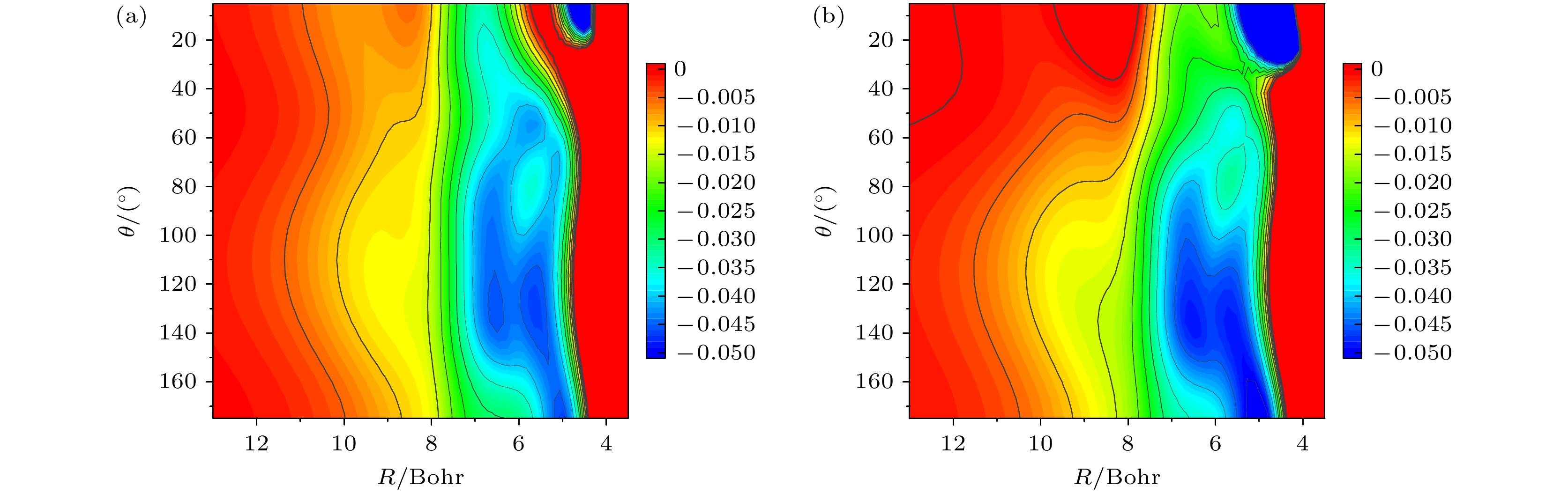


 DownLoad:
DownLoad:





