When biomolecules interact with high-energy particles and rays, they are directly ionized or dissociated, then a large number of low-energy electrons are formed as secondary particles. These low-energy electrons will attach to biomolecules, and trigger off the secondary dissociation, forming free radicals and ions with high reactivity, which can damage the structure and function of the biomolecule and cause irreversible radiation damage to the biomolecule. It is important to study the low-energy dissociative electron attachment (DEA) process of biomolecules for understanding radiation damage to biological organisms. Currently, the theoretical studies of DEA have mainly focused on the bound states of negative ions and the types of resonances in the dissociation process. The dissociation process is well described by quantum computational method, but the diversity and complexity of dissociation channels present in the dissociation process of 2-thiouracil molecule also pose a great computational challenge to these methods. In addition, the quantum computational methods are not ideal for dealing with the discrete states of chemical bonds and the problem of continuity coupling of electrons. The dissociation dynamics of biomolecules mainly results from ionization and electron attachment.
Ab initiomolecular dynamics simulation can reasonably describe these processes. In light of these considerations,
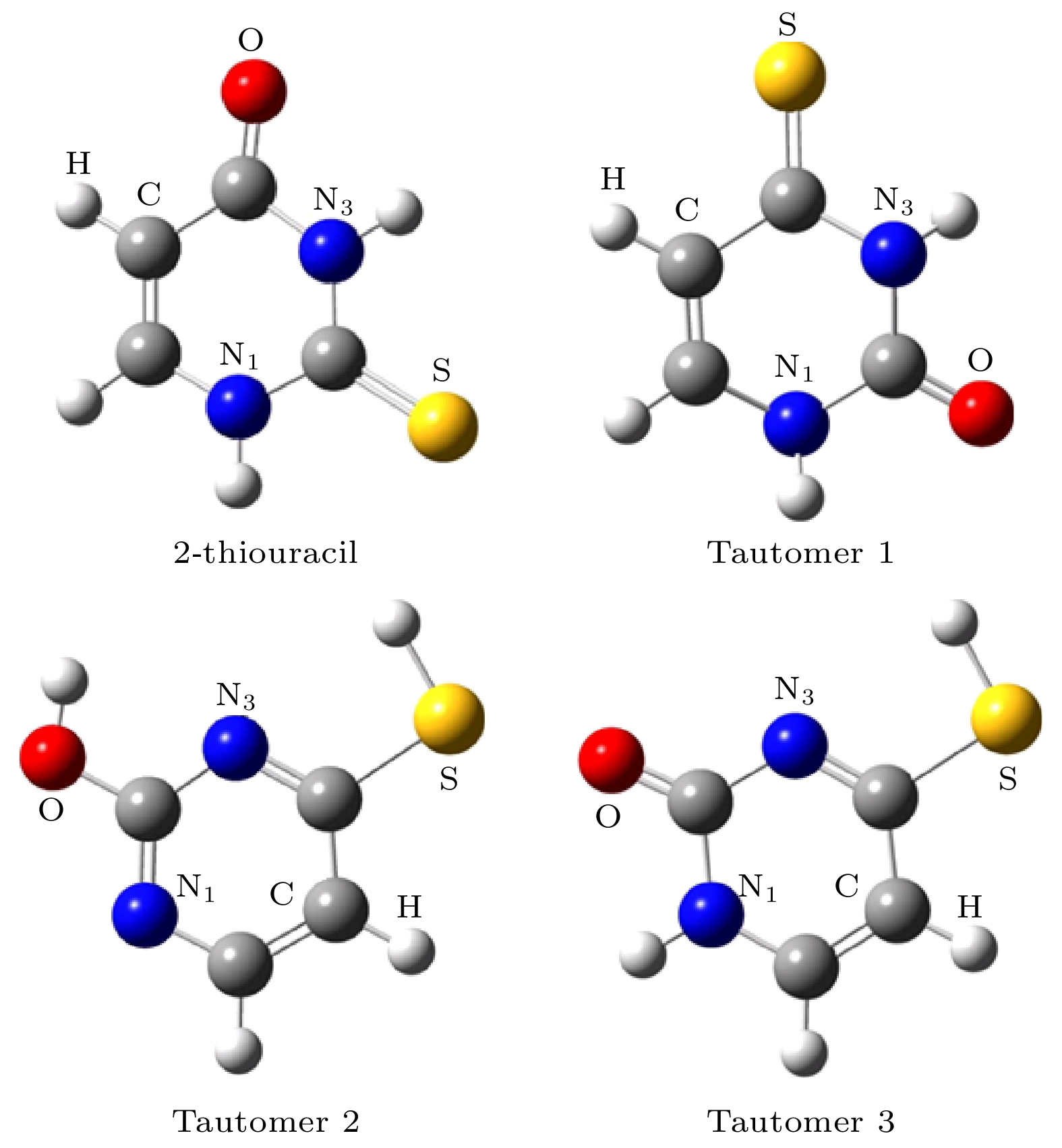
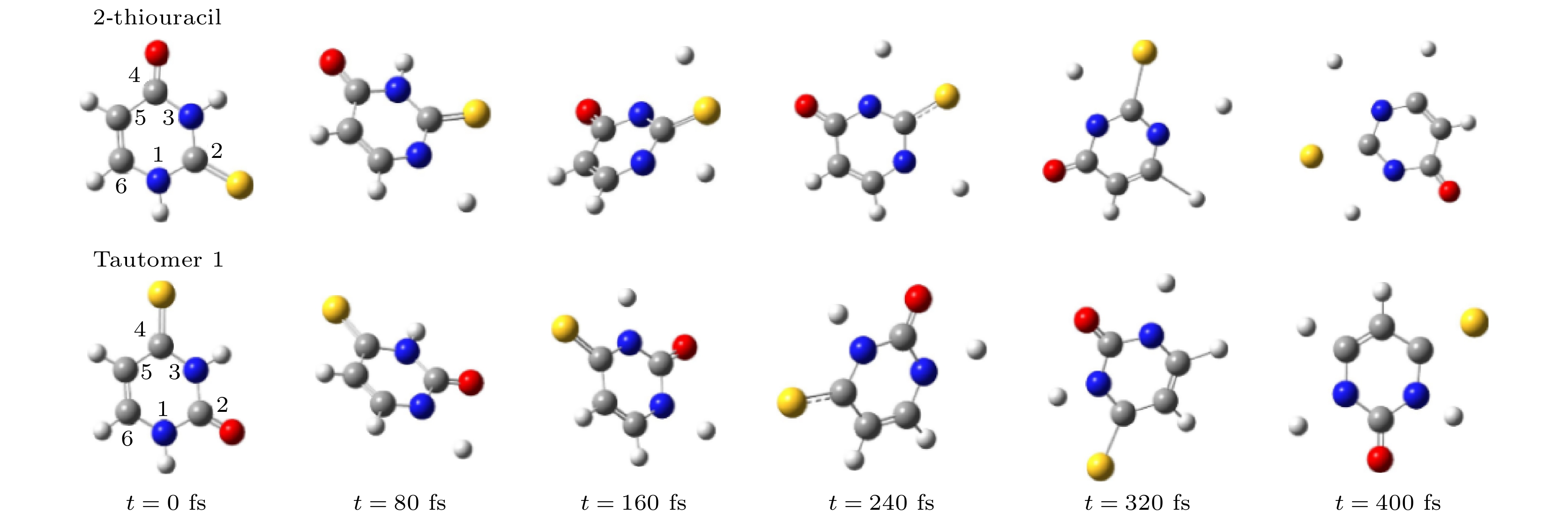
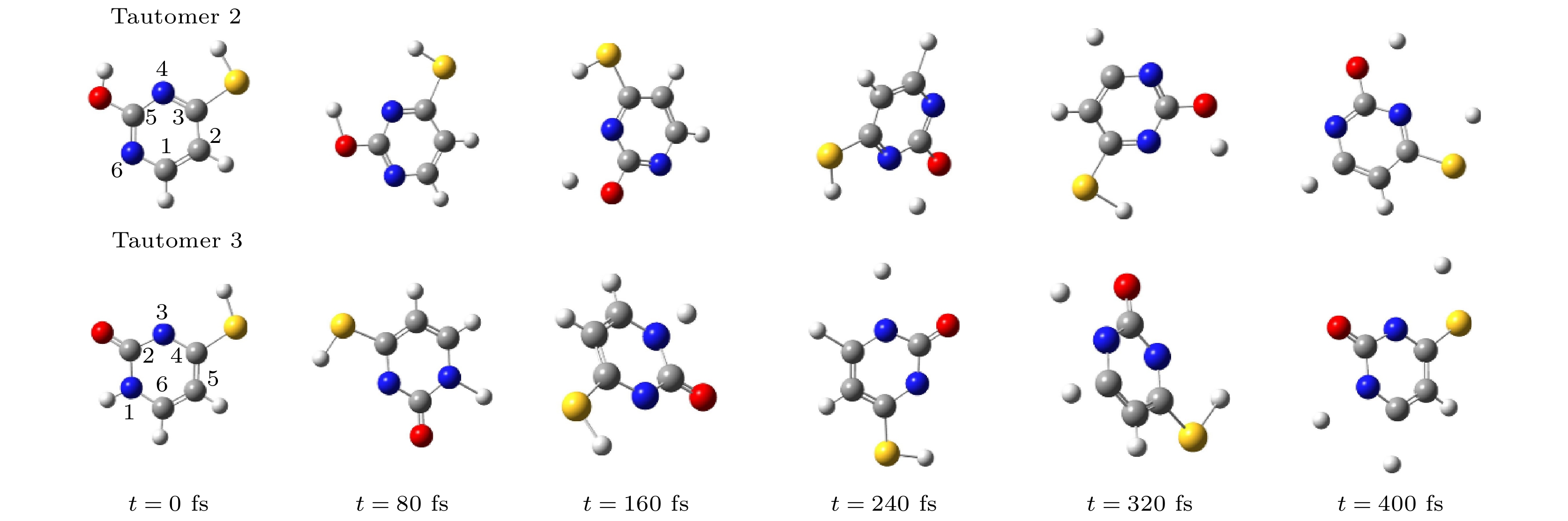
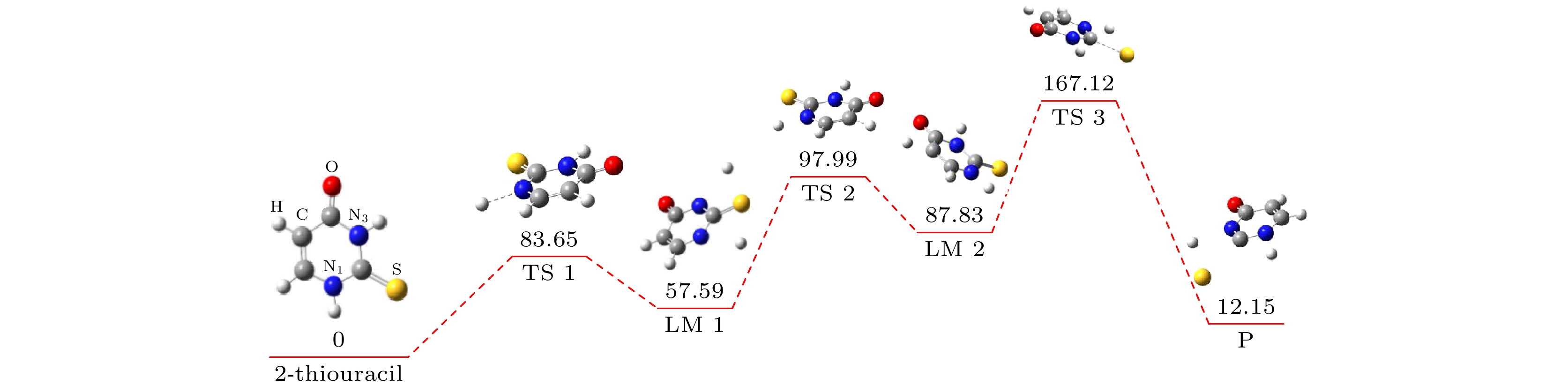
ab initiomolecular dynamics simulation is used in this work to study dynamic variation process in DEA. The low-energy electron dissociative attachment to 2-thiouracil in the gas phase is studied by using the Born-Oppenheimer molecular dynamics model combined with density functional theory. It is found that an important dehydrogenation phenomenon of 2-thiouracil and its tautomers occurs in the DEA process, and that the N—H and C—H bond are broken at specific locations. Due to the loss of hydrogen atoms at the N and C sites, the closed-shell dehydrogenated negative ion (TU-H)
–forms, which is the most important negative ion fragments in the dissociation process. The potential energy curves, the bond dissociation energy and the electron affinity energy of the broken bond show that the N—H bond is the most likely to break, indicating the formation of the negative ion (TU-H)
–mainly comes from the breaking of N—H bond. The theoretical calculations in this work are in good agreement with the available experimental results, indicating that the chosen calculation method is fully reliable. The BOMD simulations can not only dynamically recover the process of dissociative attachment of low-energy electrons to 2-thiouracil, but also more importantly provide an insight into the mechanisms of dehydrogenation and dissociation channels of 2-thiouracil molecules in DEA process.












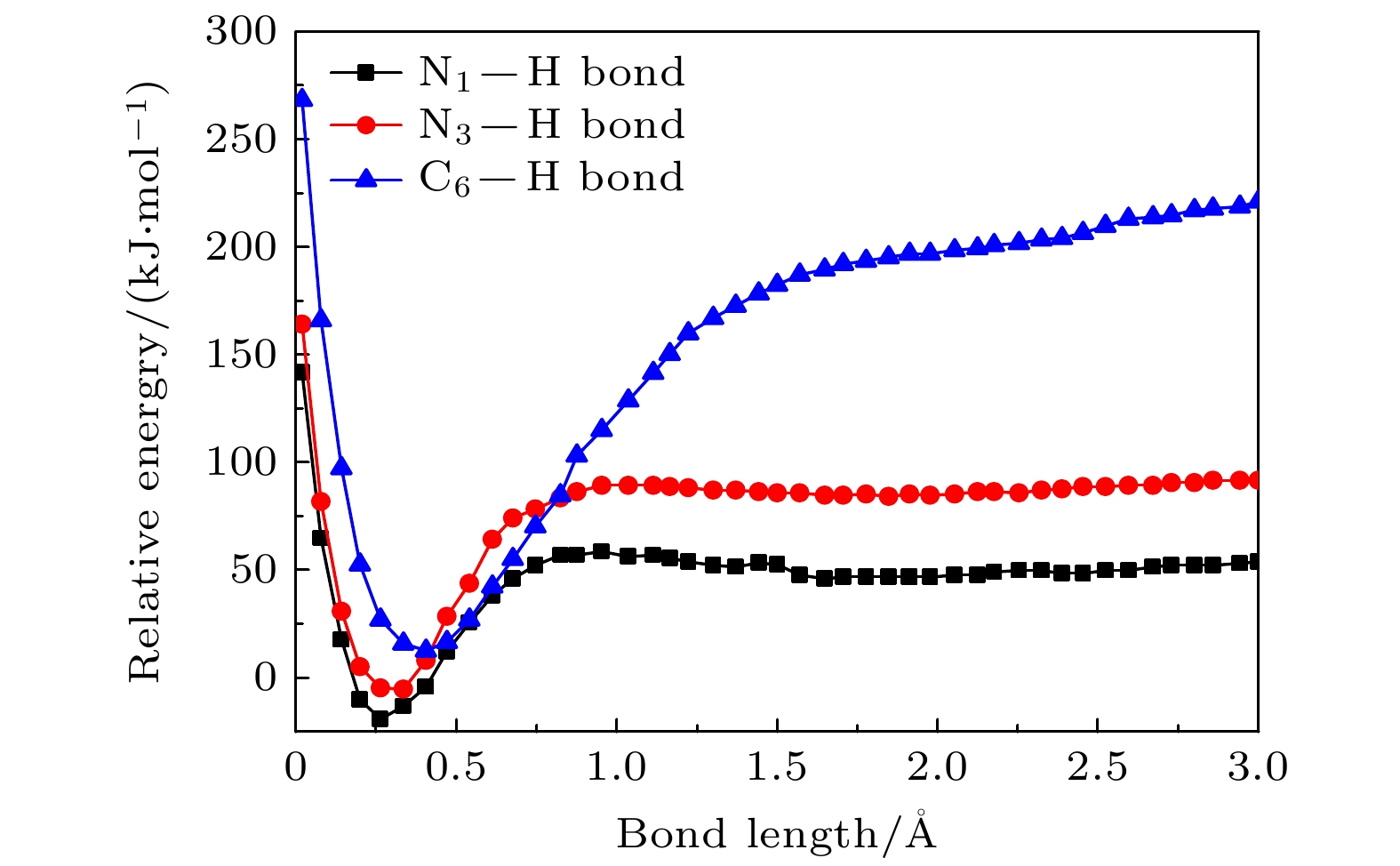
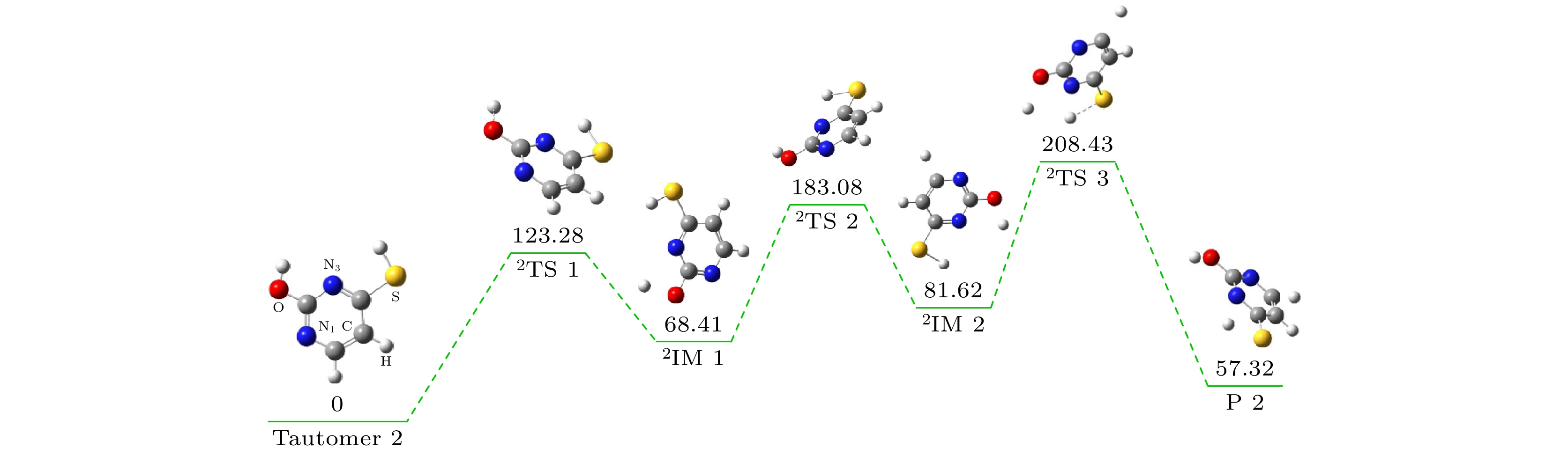
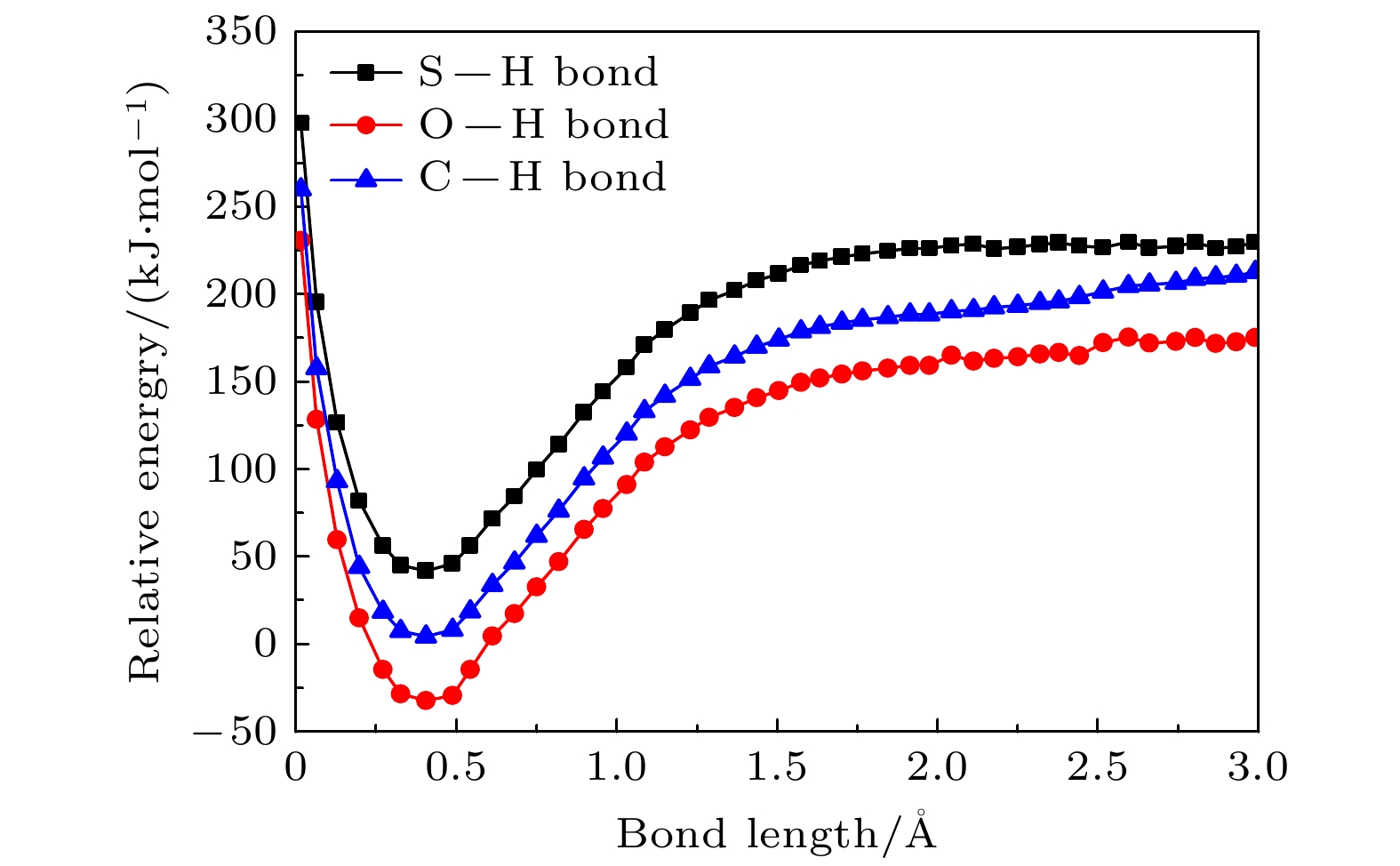
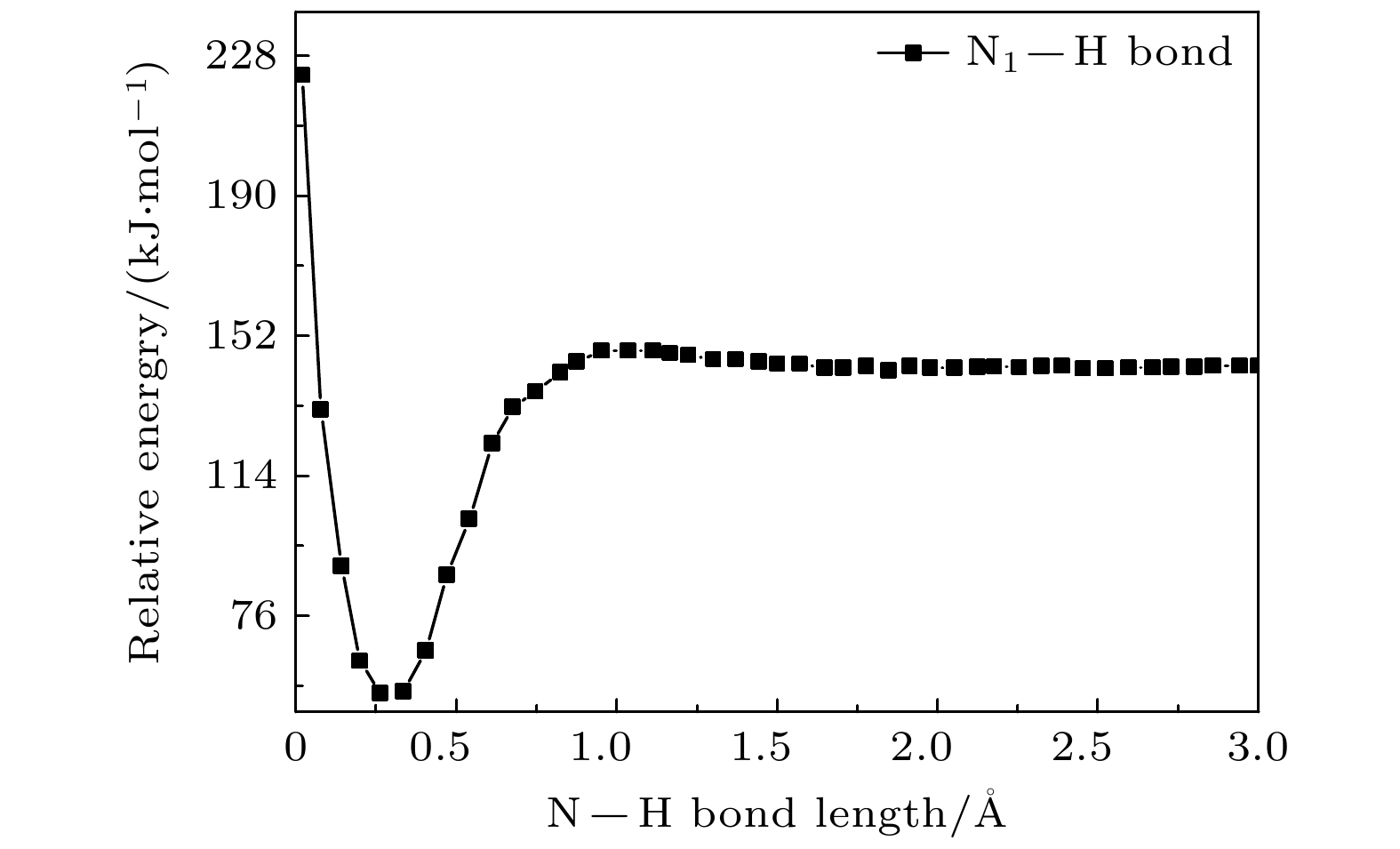
 DownLoad:
CSV
DownLoad:
CSV


 DownLoad:
DownLoad:






