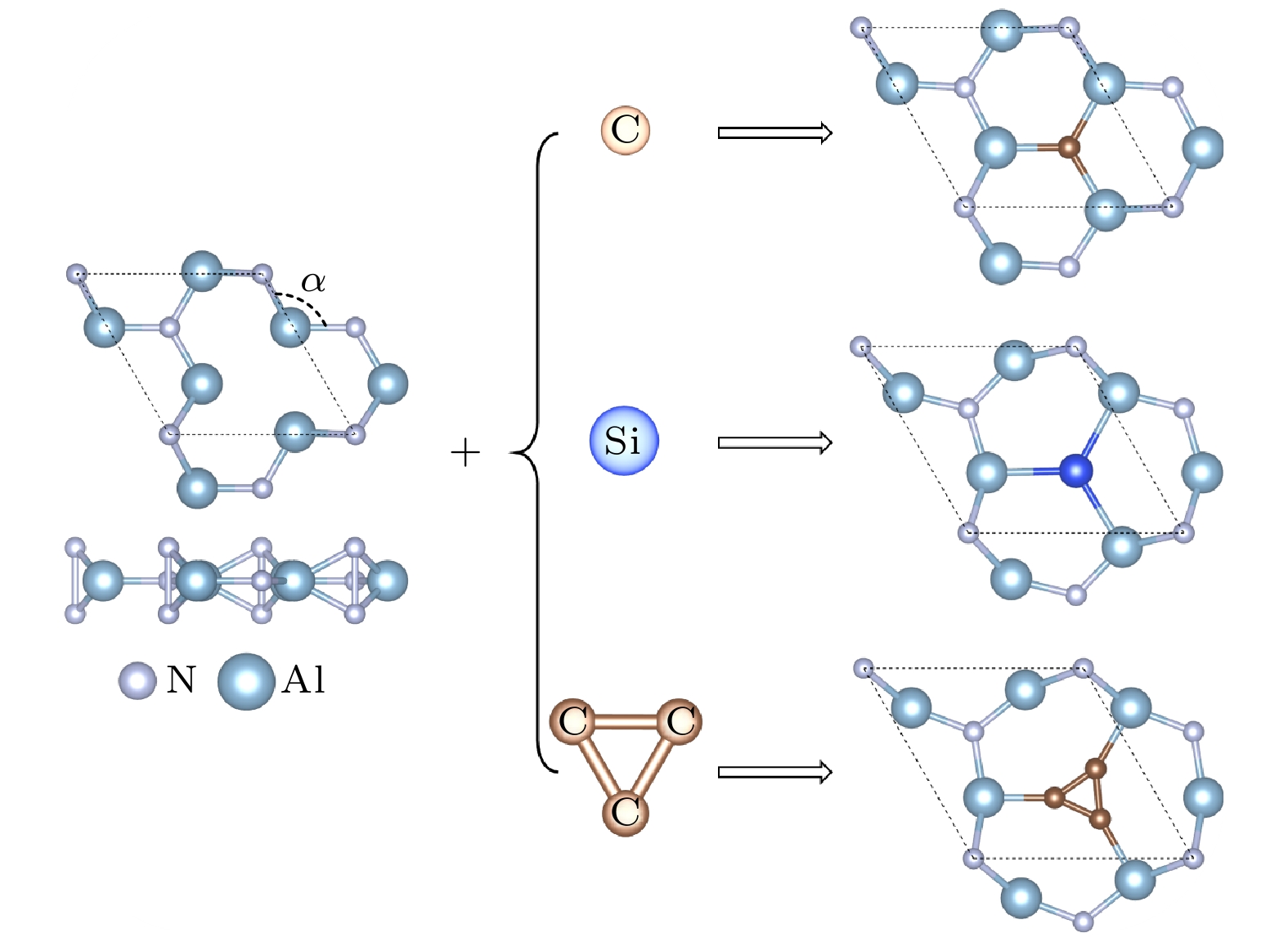
Aluminum nitride (AlN) is of paramount importance in developing electronic devices because of excellent stability and thermal transport performance. However, lack of novel materials which can provide colorful physical and chemical properties seriously hinders further digging out application potential. In this work, we perform an evolutionary structural search based on first-principles calculation and verify the dynamic and thermal dynamic stability of porous buckled AlN and
X-AlN (
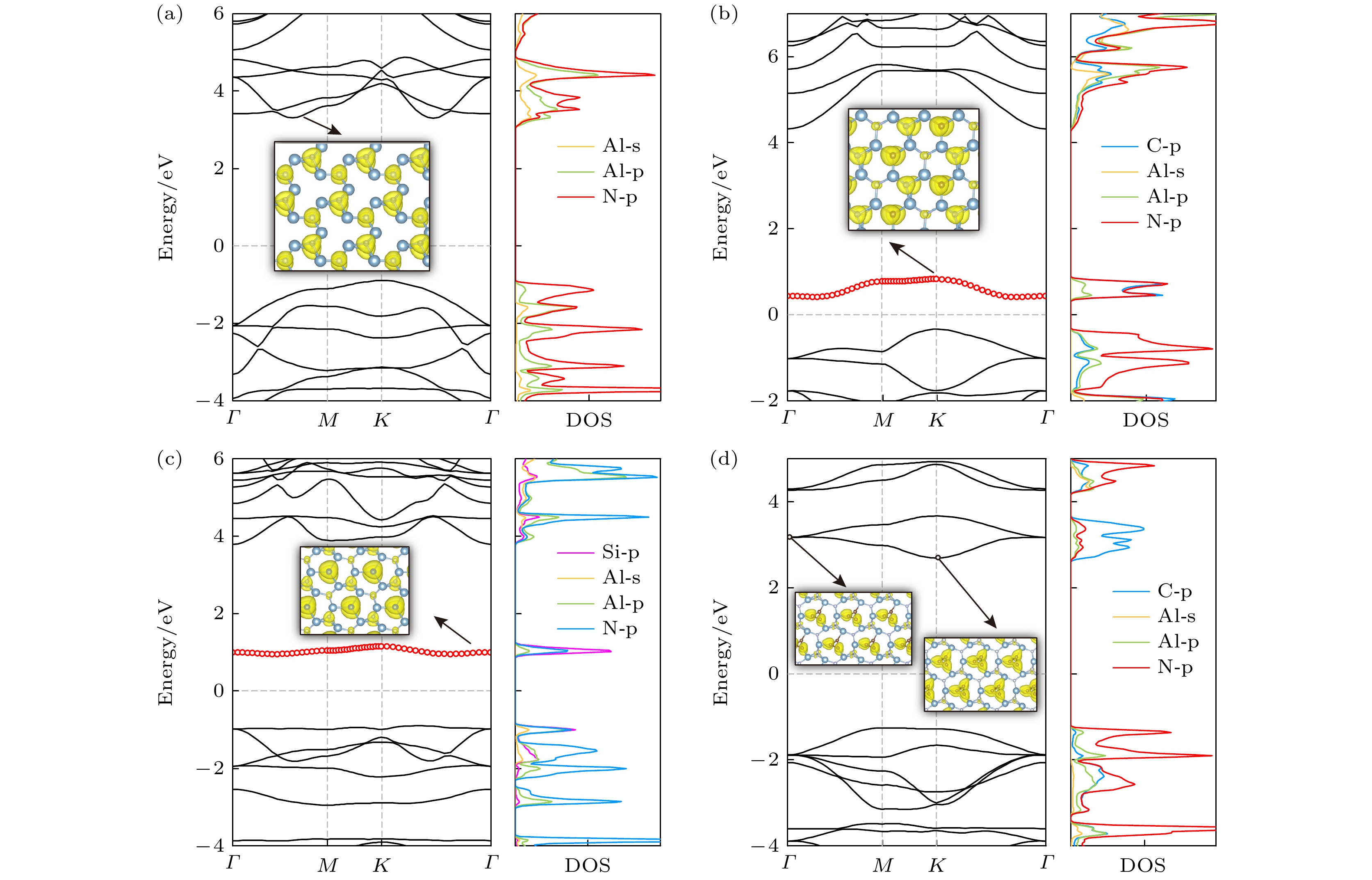
X= C, Si, TC) structural system, which constructs by introducing C, Si atoms and triangular carbon (TC) into the porous vacancy of AlN, by calculating phonon spectra and first-principles molecular dynamic simulations. Structural deformation becomes gradually serious with the increase of structural unit size and significantly influences structural, electronic, and thermal transport properties. Firstly, we point out that a flat energy band appears around the Fermi level in C-AlN and Si-AlN because of weak interatomic interaction between C/Si and the neighbor Al atoms. Unoccupied C-/Si-p
zand Al-p
zdo not form
$ {\rm{\pi }} $

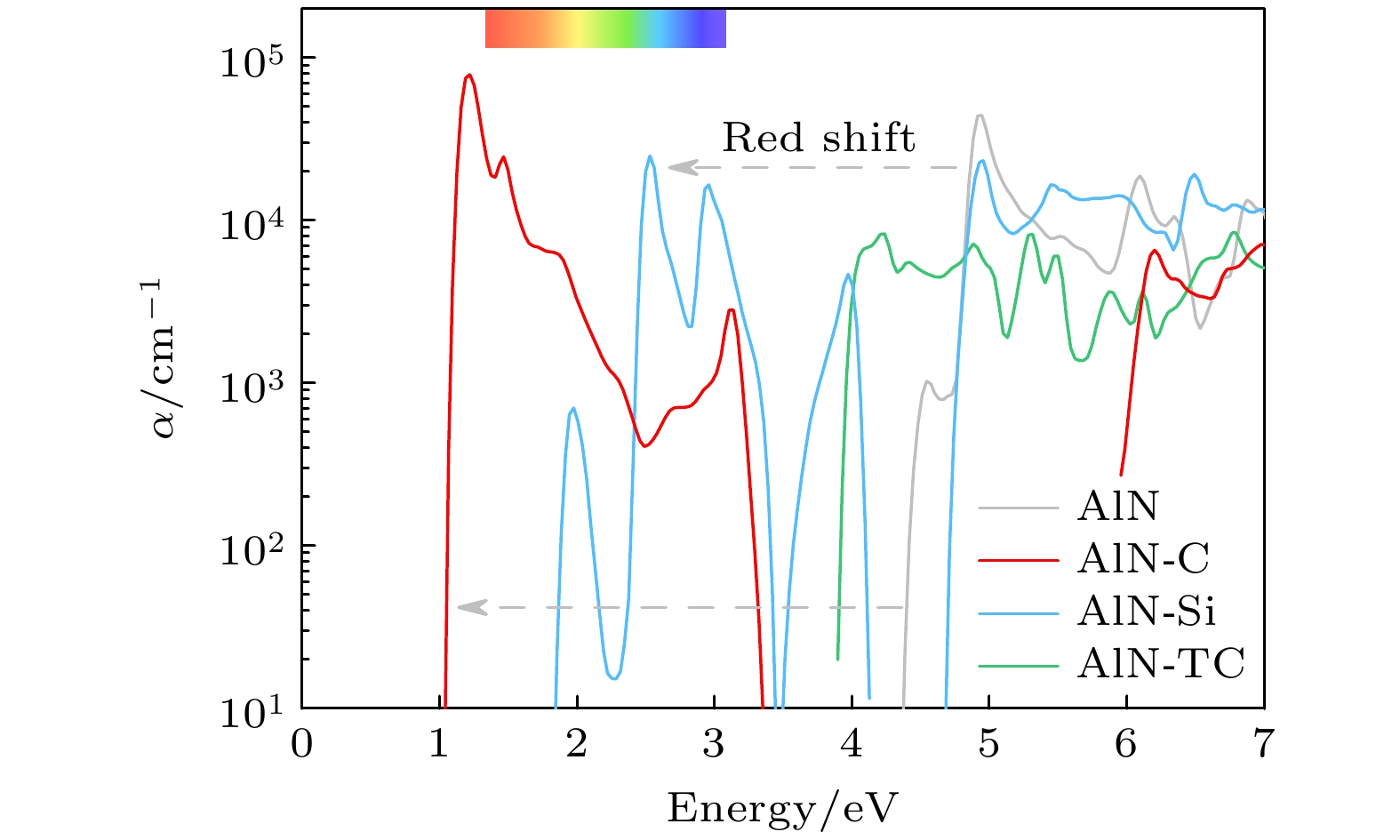
bond and only a localized flat band near Fermi level arises, and thus the absorption peaks of structures are enhanced and the red shift occurs. Bonding state of
$ {\rm{\pi }} $
bond from hybridized C-p
zorbitals in triangular carbon of TC-AlN lowers the energy of conduction band at
Kpoint in the first Brillouin zone and the corresponding antibonding state raises the band at
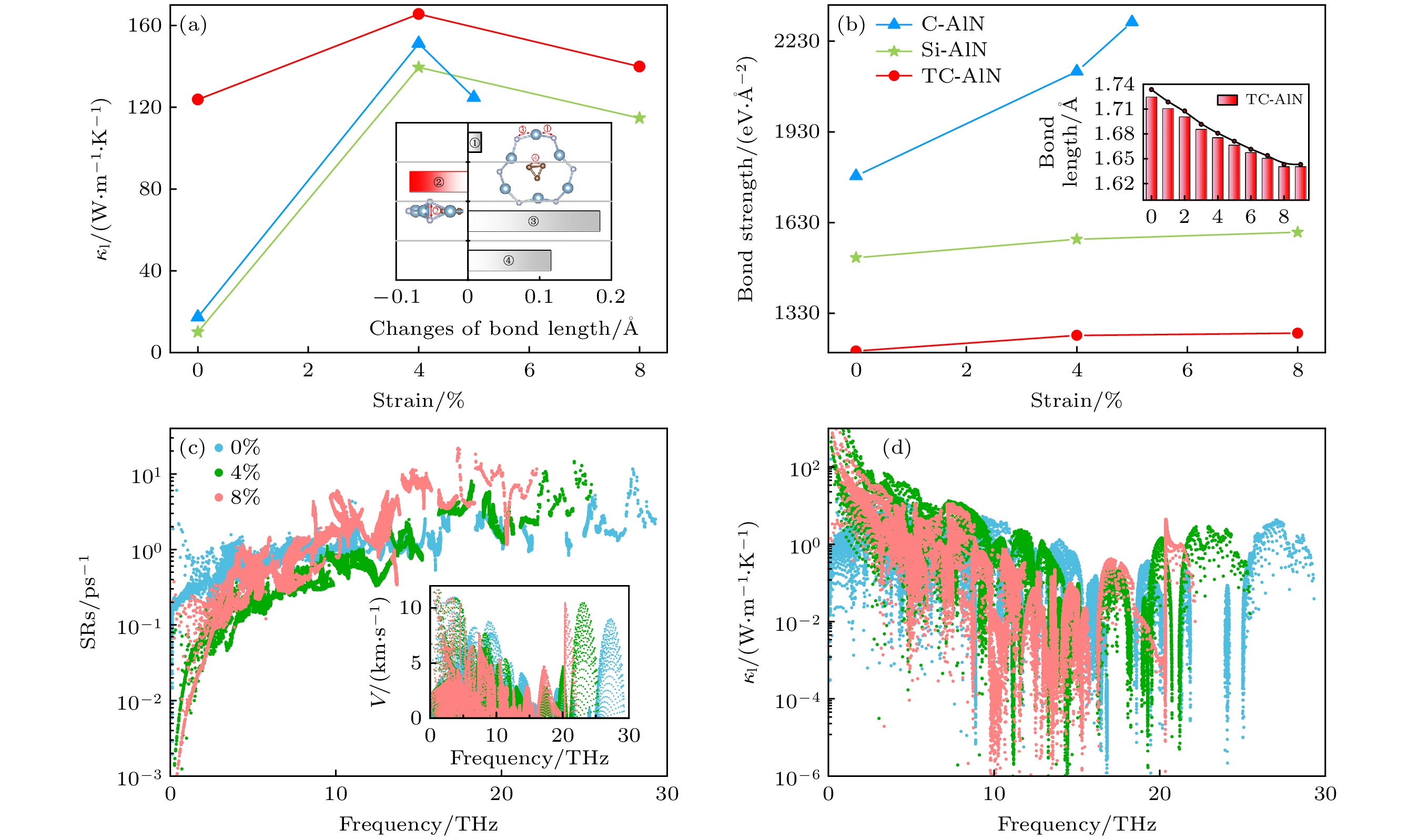
Γ, therefore transition from indirect bandgap of AlN to direct bandgap of TC-AlN appears. Secondly, porous buckled AlN shows the lowest thermal conductivity due to asymmetric Al—N bonds around the porous vacancy and vertically stacked N—N bonds. Introduced C and Si atoms both reduce structural anharmonicity, while the former has a relatively small distortion, and so it has a higher thermal conductivity. Triangular carbon in TC-AlN hinders phonon scattering between FA mode and other phonon modes and has the weakest anharmonicity because of the strongest bond strength, and obtains the highest thermal transport performance. Finally, we unveil the physical mechanism of anomalous thermal conductivity in
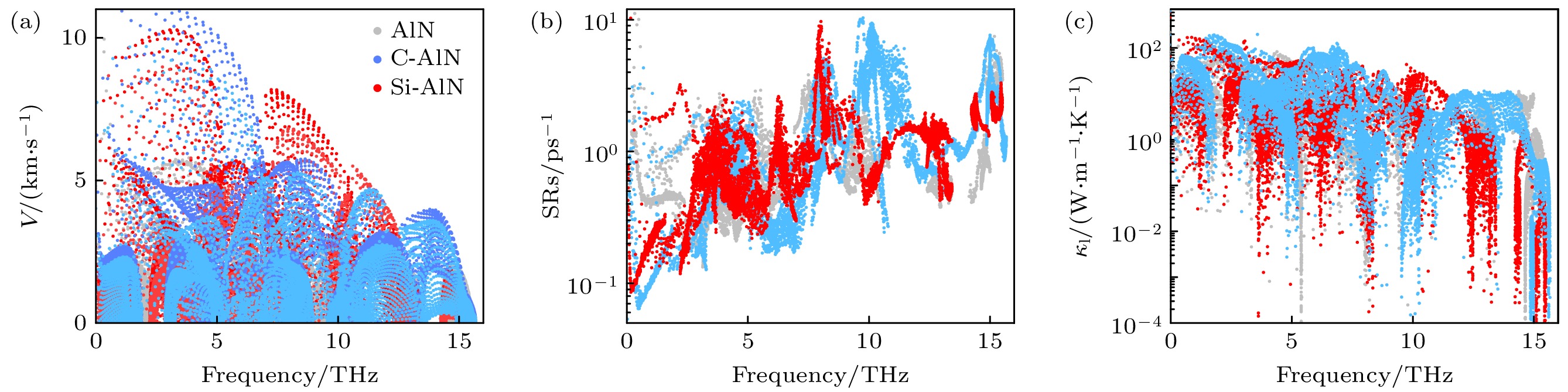
X-AlN system by modulating the biaxial tensile strain. Enhanced vertical N—N bonds dominate thermal transport due to its weaker anharmonicity with a slightly strain, and when tensile strain is above the 4%, soften phonon modes reduce phonon velocity and thus hinders the thermal transport process. Therefore, occurs the anomalous thermal transport behavior, i.e. thermal conductivity first rises and then drops with applied biaxial strain increasing. Our work paves the way for modulating two-dimensional AlN performance and provides a new insight for designing promising novel two-dimensional semiconductors.













 DownLoad:
CSV
DownLoad:
CSV
 096301-20230116补充材料.pdf
096301-20230116补充材料.pdf





 DownLoad:
DownLoad:




