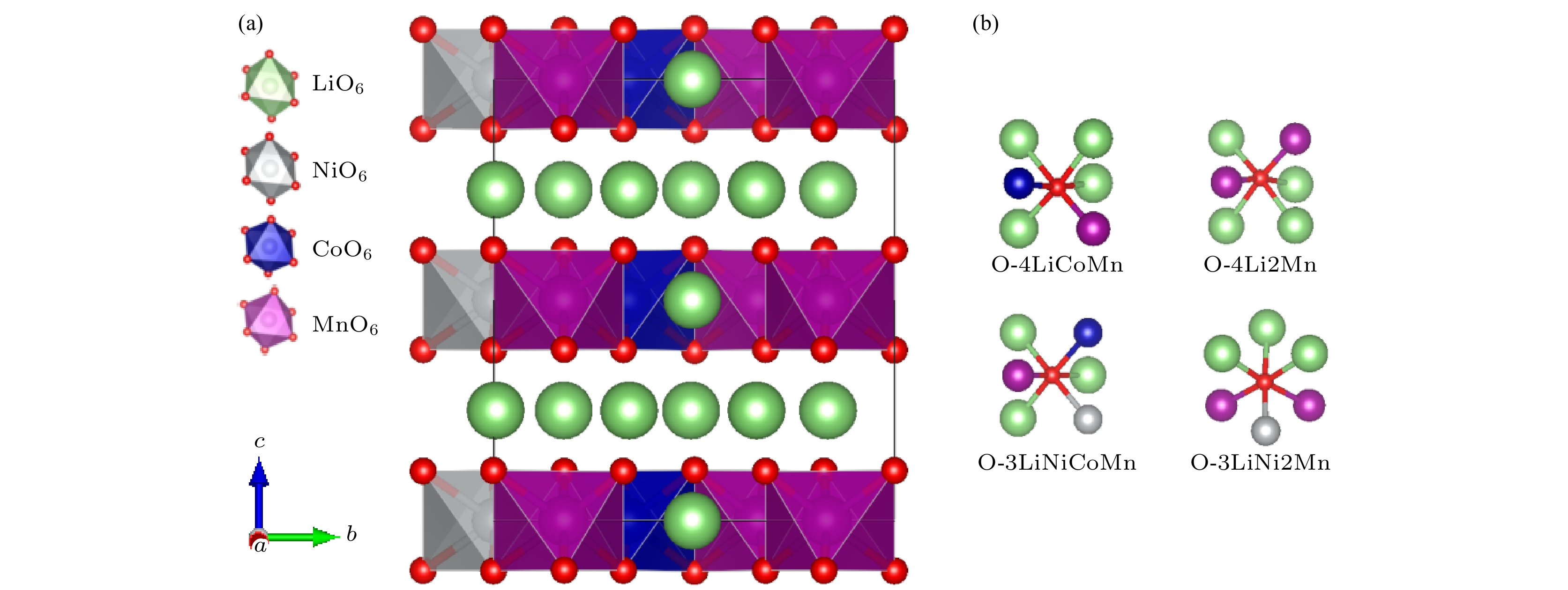
Using the first-principles method based on the density functional theory, the oxygen vacancy formations in the lithium-rich manganese-based ternary cathode material Li
1.167Ni
0.167Co
0.167Mn
0.5O
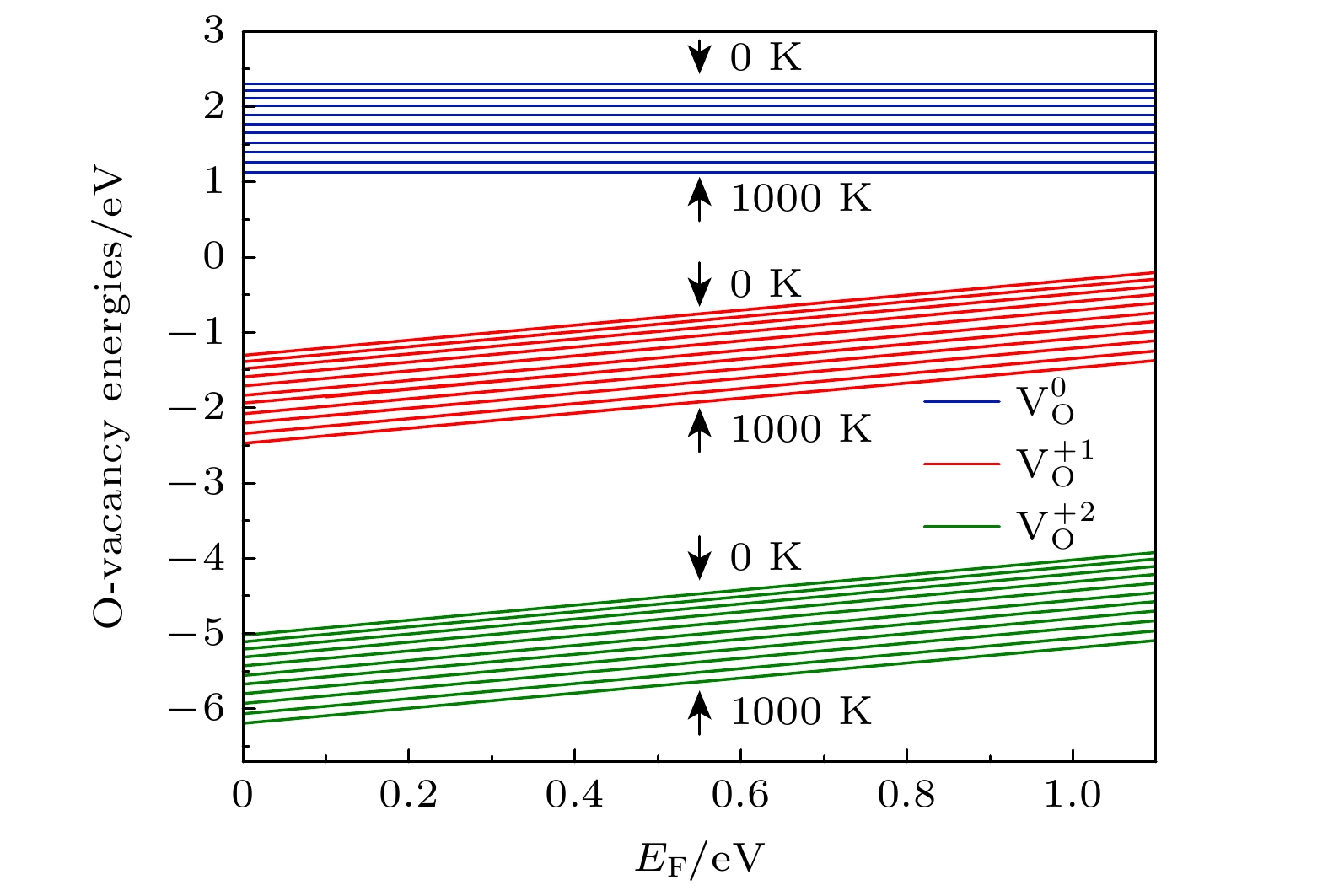
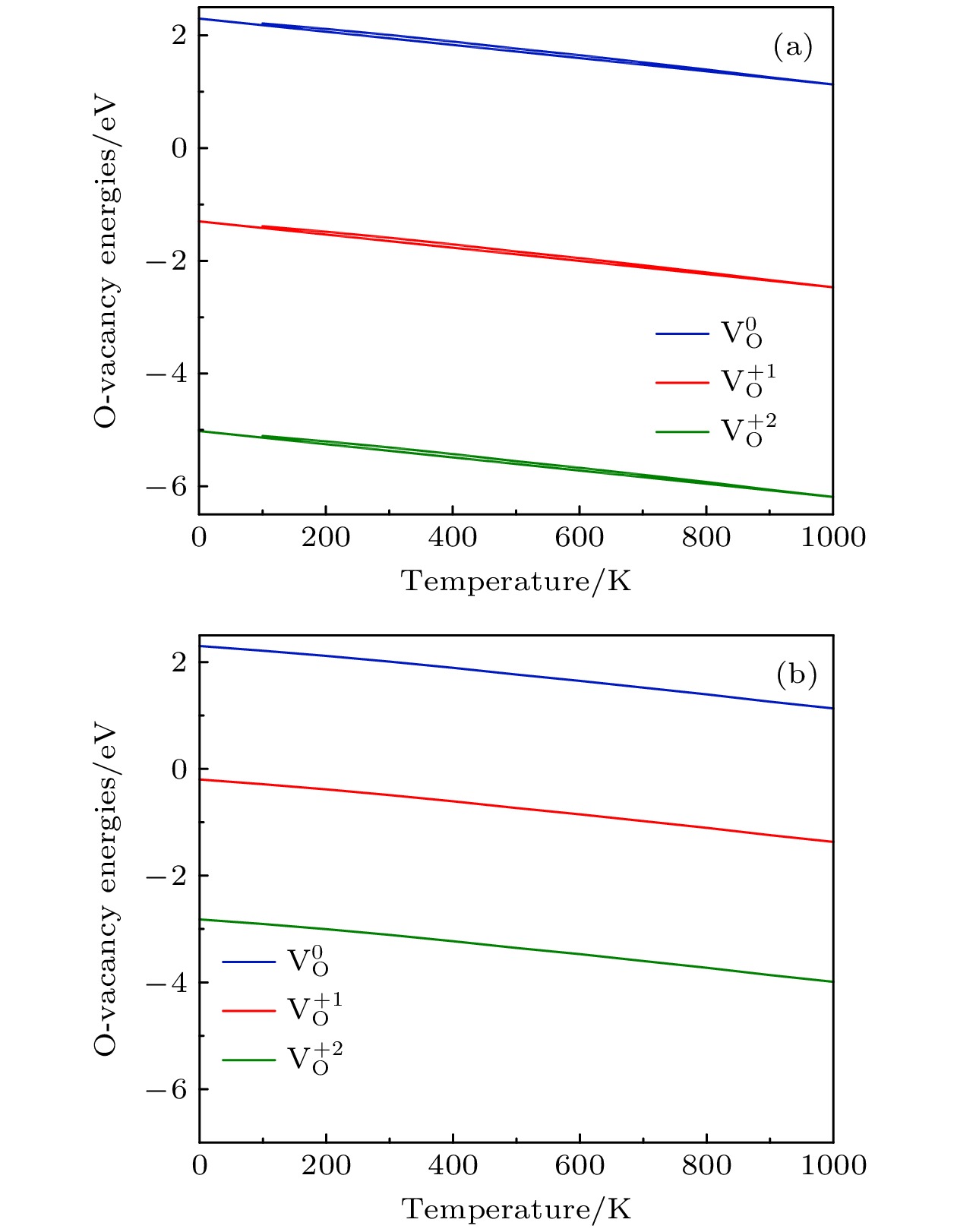
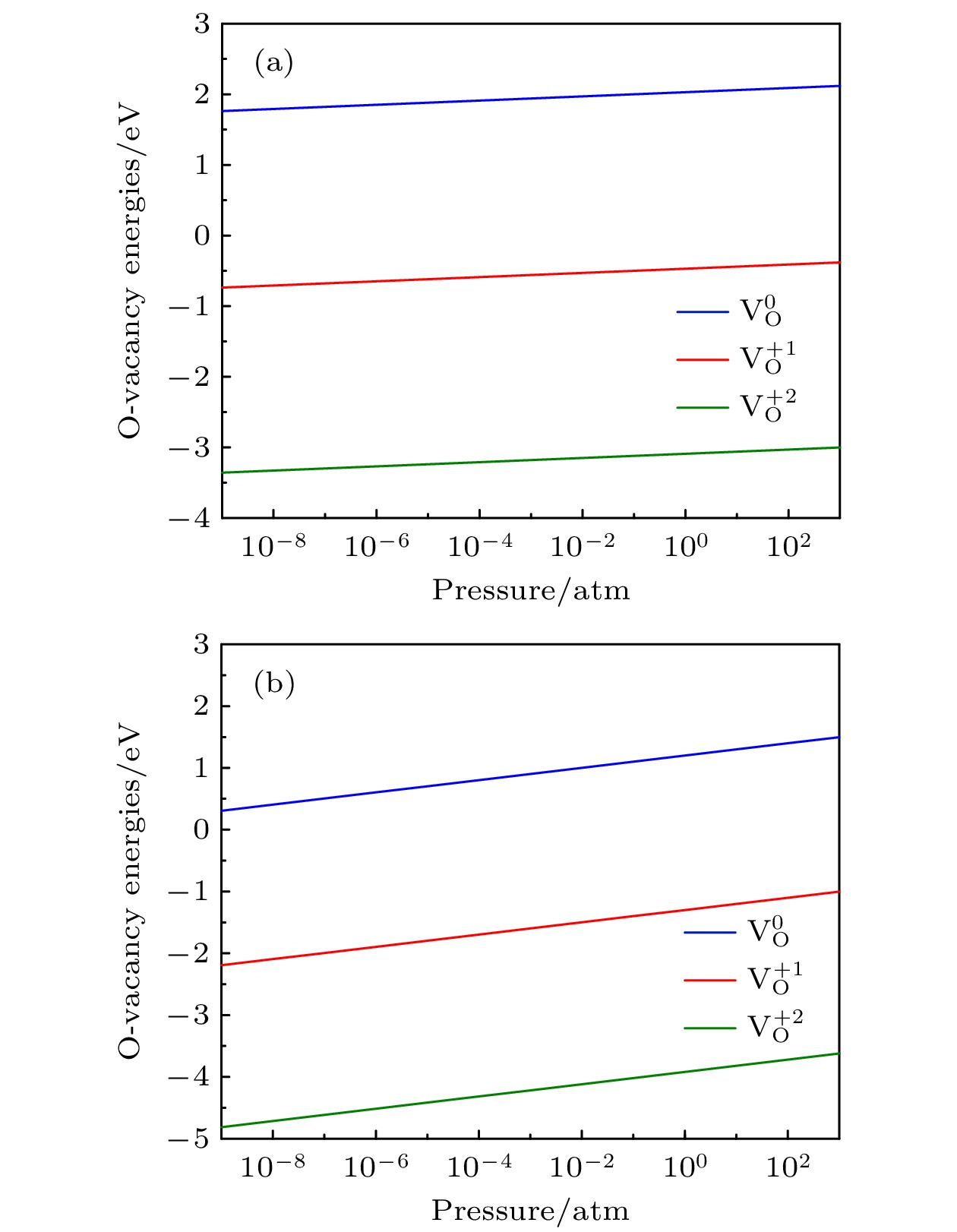
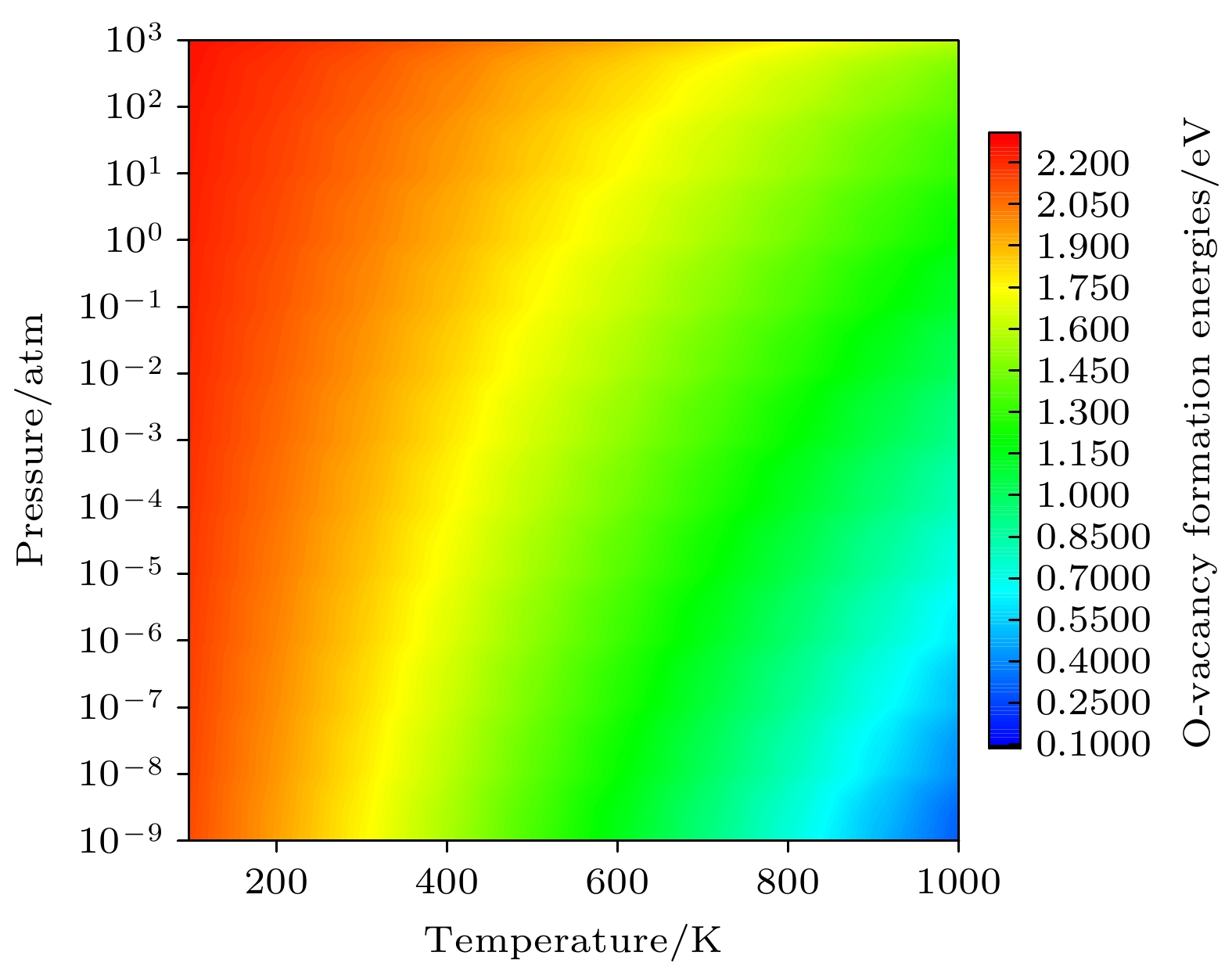
2are calculated. The changes of oxygen vacancy formation energy with temperature, oxygen partial pressure and point defects in the material are discussed, meanwhile, the effect of oxygen vacancies on the capacity is also discussed. The calculation results show that the increase of temperature and the decrease of oxygen partial pressure can lead the formation energy of an oxygen vacancy to decline. For the charged oxygen vacancies (
$ {\mathrm{V}}_{\mathrm{O}}^{+1} $

,
$ {\mathrm{V}}_{\mathrm{O}}^{+2} $

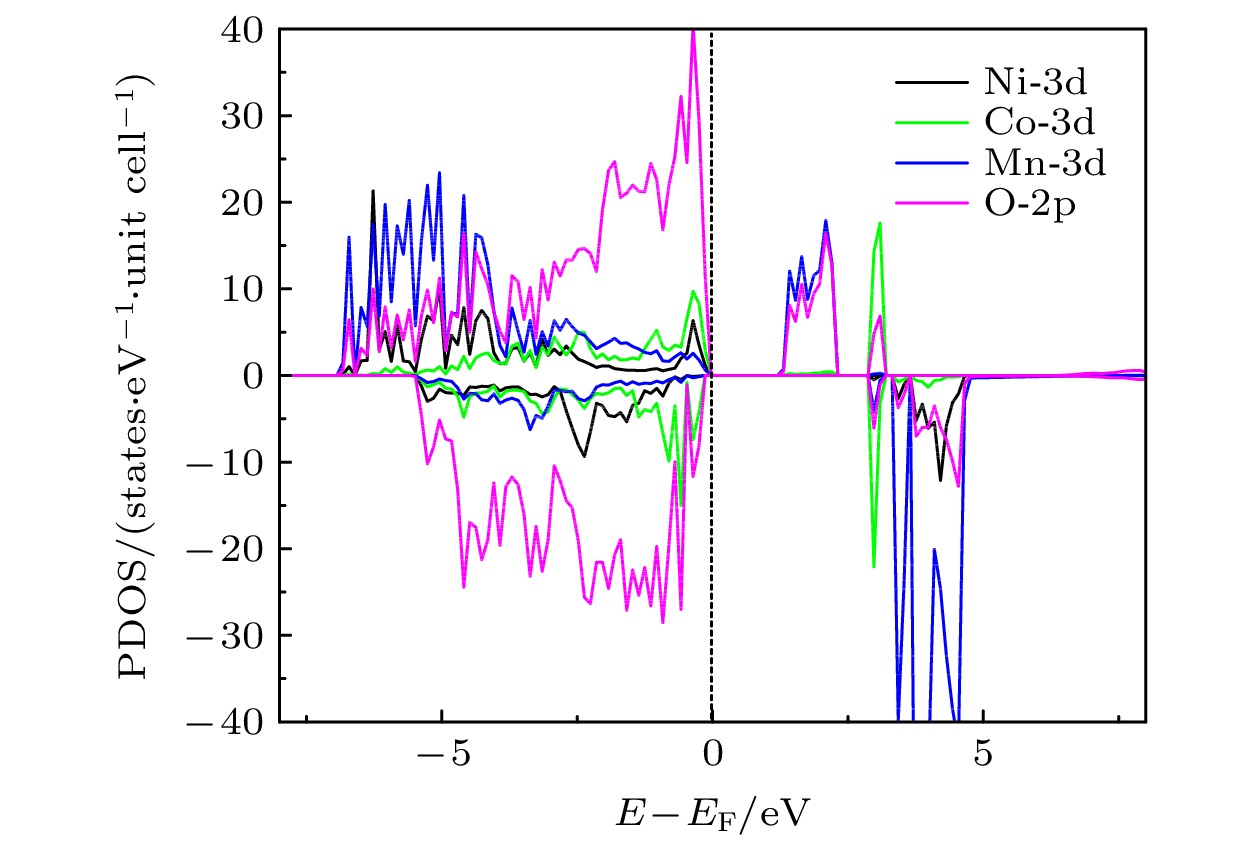
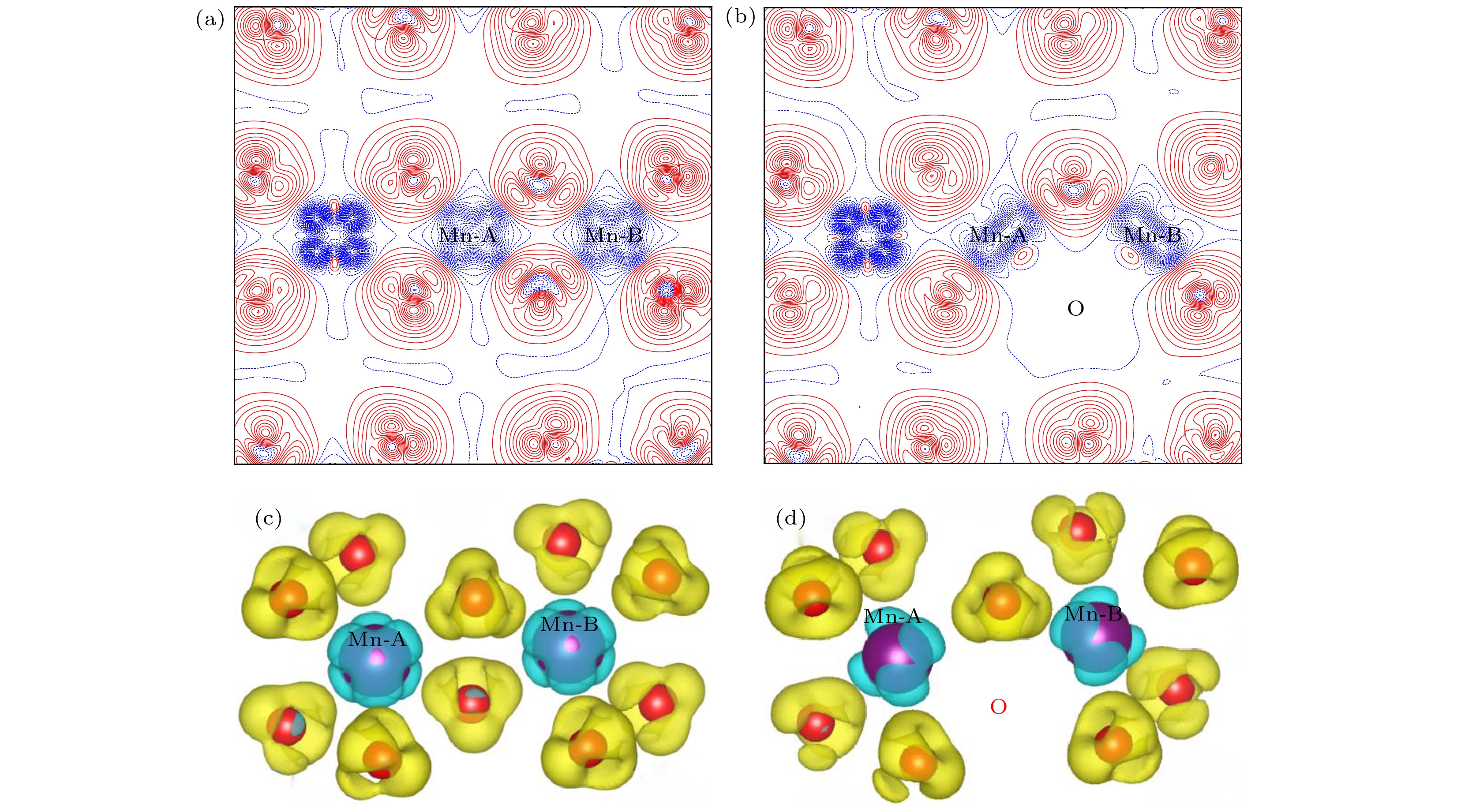
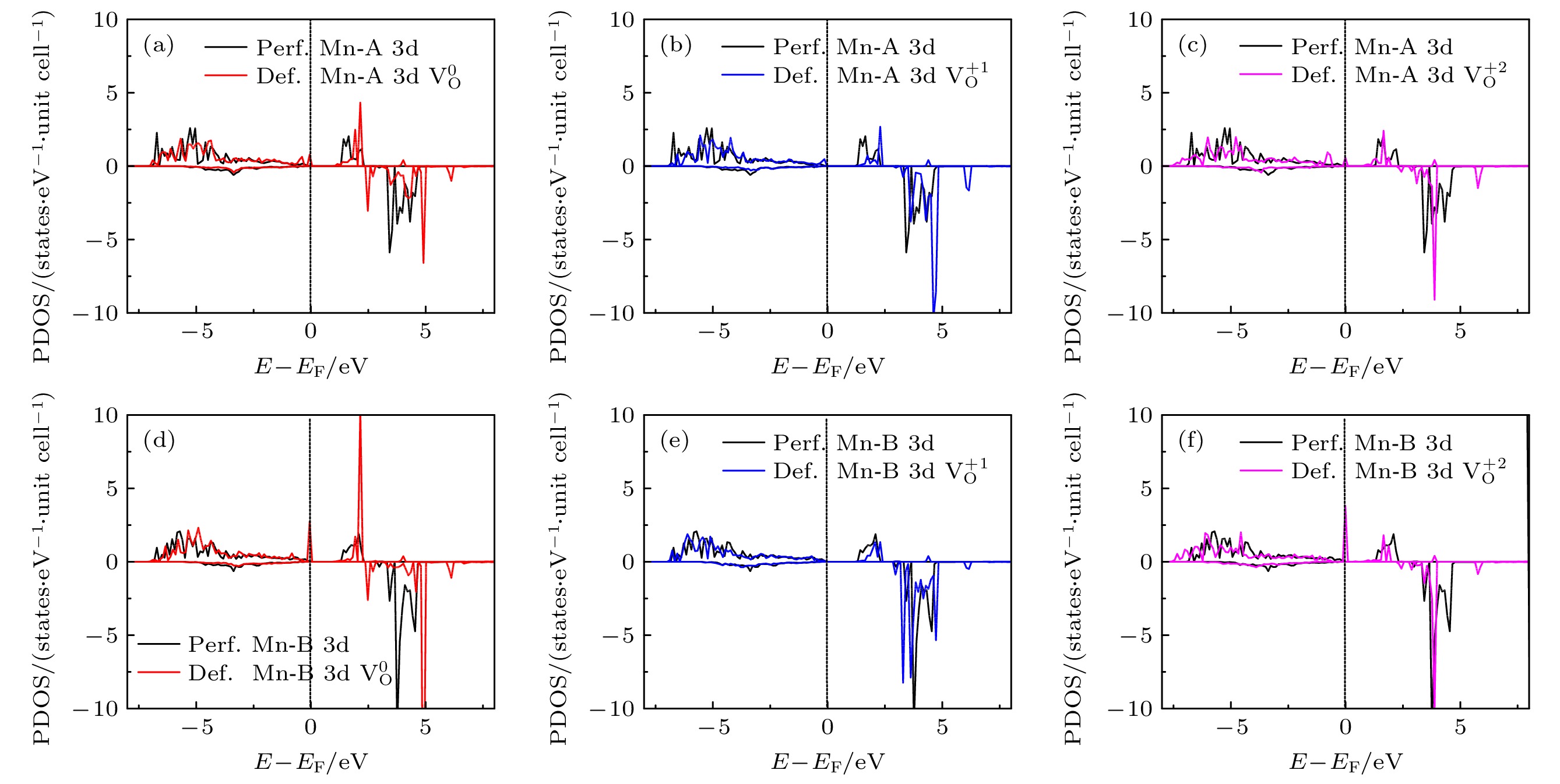
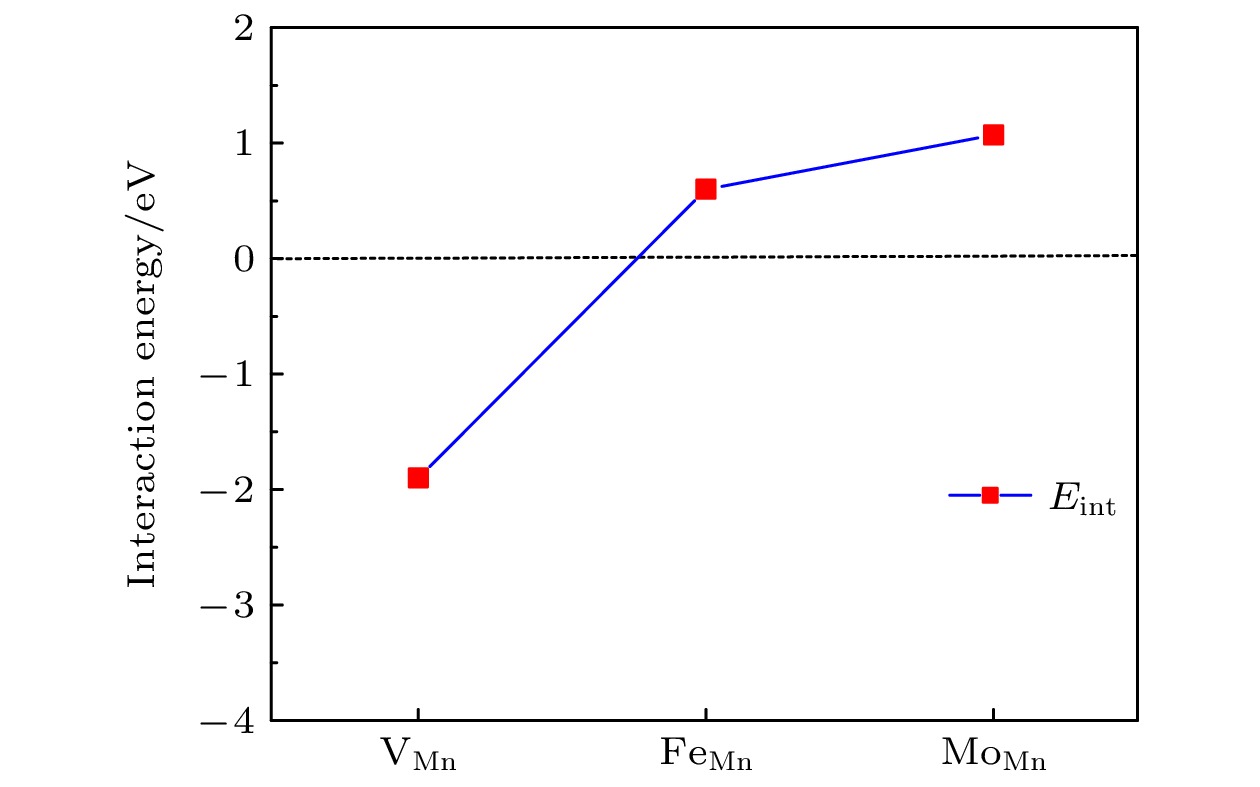
), the formation energy of an O-vacancy increases with Fermi level increasing. It is also found that the presence of an oxygen vacancy will trigger off a very local charge density redistributions, mainly around the neighboring Mn ions next to the O-vacancy. Furthermore, the effects of point defects, including cation vacancies and substitutional defects in the vicinity of the O-vacancy, on the formation energy of O-vacancy are also calculated. The results show that the presence of Mn vacancy near the O-vacancy is beneficial to the formation of the O-vacancy. In addition, the formation of oxygen vacancy is suppressed when the Mn atoms near the O-vacancy are substituted by the Mo or Fe atoms.












 DownLoad:
CSV
DownLoad:
CSV




 DownLoad:
DownLoad:







