Quantum calculation of molecular vibrational frequency is important in investigating infrared spectrum and Raman spectrum. In this work, a low computational cost method of calculating the quantum chemistry of vibrational frequencies for large molecules is proposed. Usually, the calculation of vibrational frequency of a molecule containing
Natoms needs to deal with the Hessian matrix, which consists of second derivatives of the 3
N-dimensional potential hypersurface, and then solve secular equations of the matrix to obtain normal vibration modes and the corresponding frequencies. Larger
Nimplies higher computational cost. Therefore, for a limited computational hardware condition, higher-level computations for large
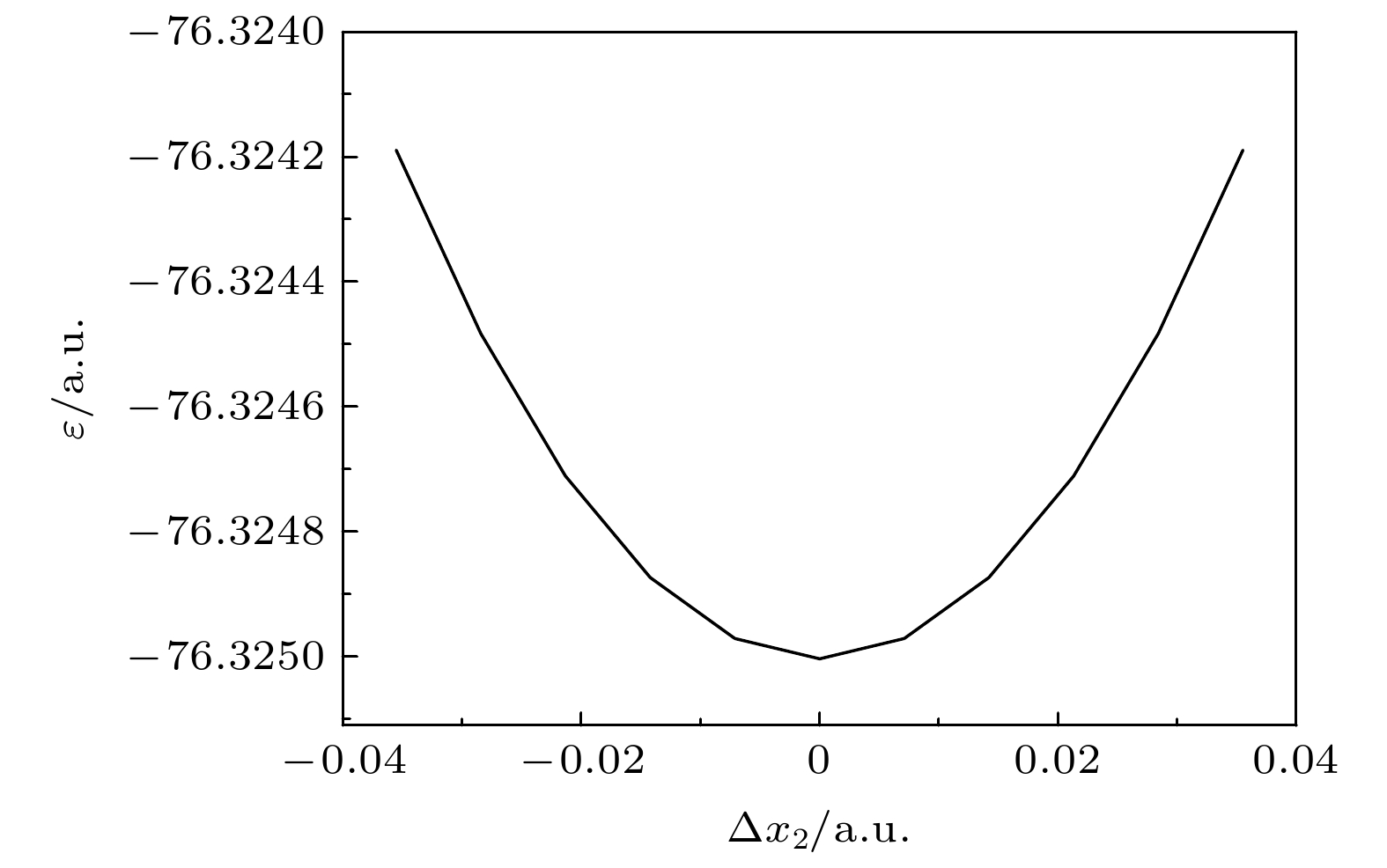
Natomic molecule’s vibrational frequencies cannot be implemented in practice. Here we solve this problem by calculating the vibrational frequency for only one vibrational mode each time instead of calculating the Hessian matrix to obtain all vibrational frequencies. When only one vibrational mode is taken into consideration, the molecular potential hypersurface can be transformed into one-dimensional curve. Hence, we can calculate the curve with high-level computational method, then deduce the expression of one-dimensional curve by using harmonic oscillating approximation and obtain the vibrational frequency by using the expression to fit the curve. It should be noted that this method is applied to vibrational modes whose vibrational coordinates can be completely determined by equilibrium geometry and the molecular symmetry and be independent of the molecular force constants. It requires that there exists no other vibrational mode with the same symmetry but with different frequencies. The lower computational cost for a one-dimensional potential curve than that for 3
N-dimensional potential hypersurface’s second derivatives permits us to use higher-level method and larger basis set for a given computational hardware condition to achieve more accurate results. In this paper we take the calculation of
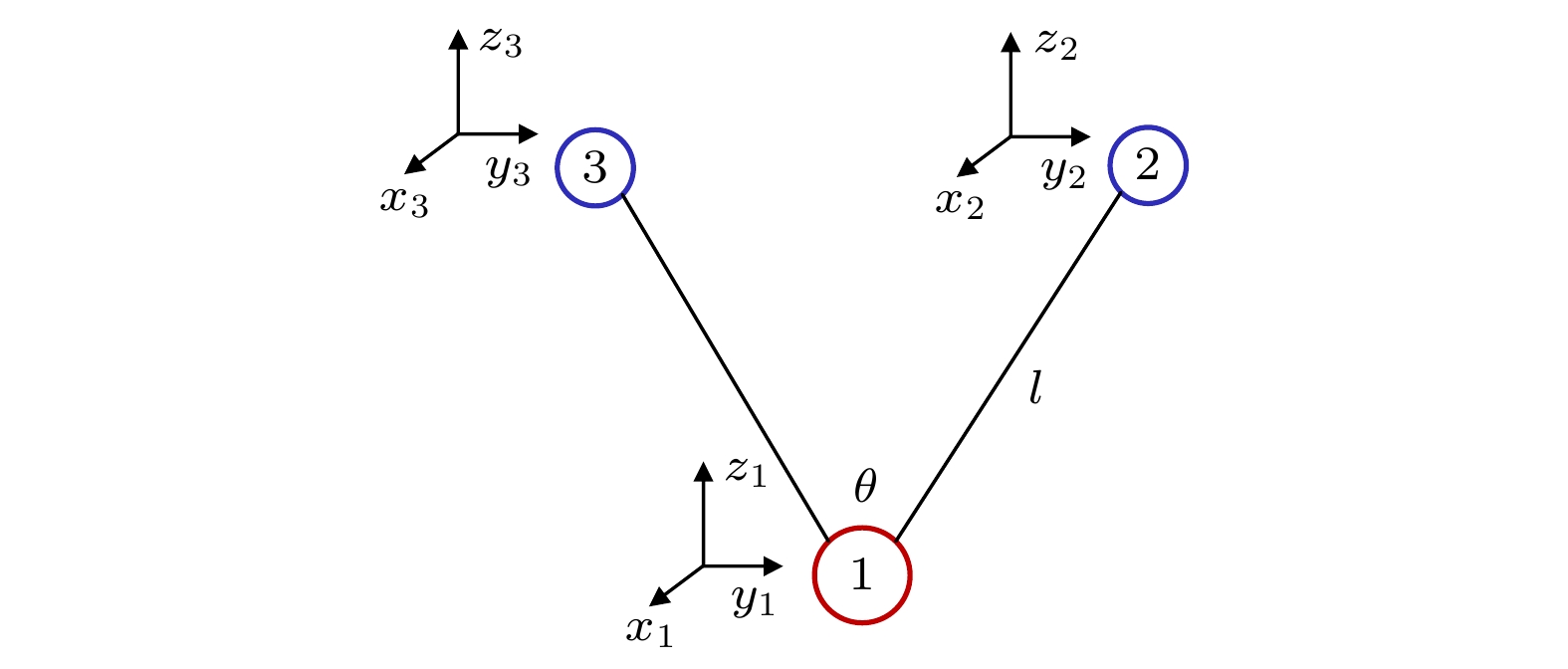
B
2vibrational frequency of water molecule for example to illustrate the feasibility of this method. Furthermore, we use this method to deal with the SF
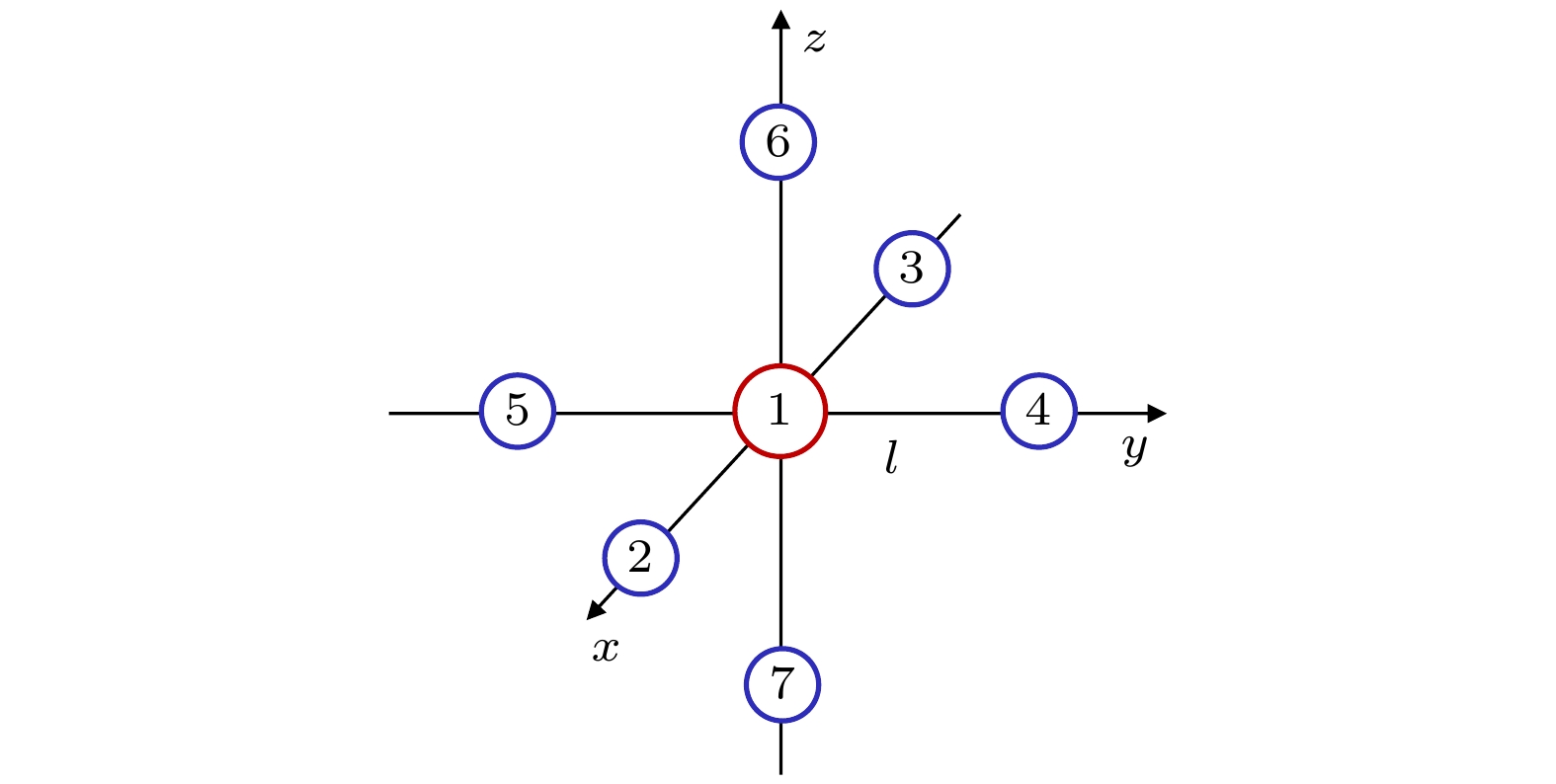
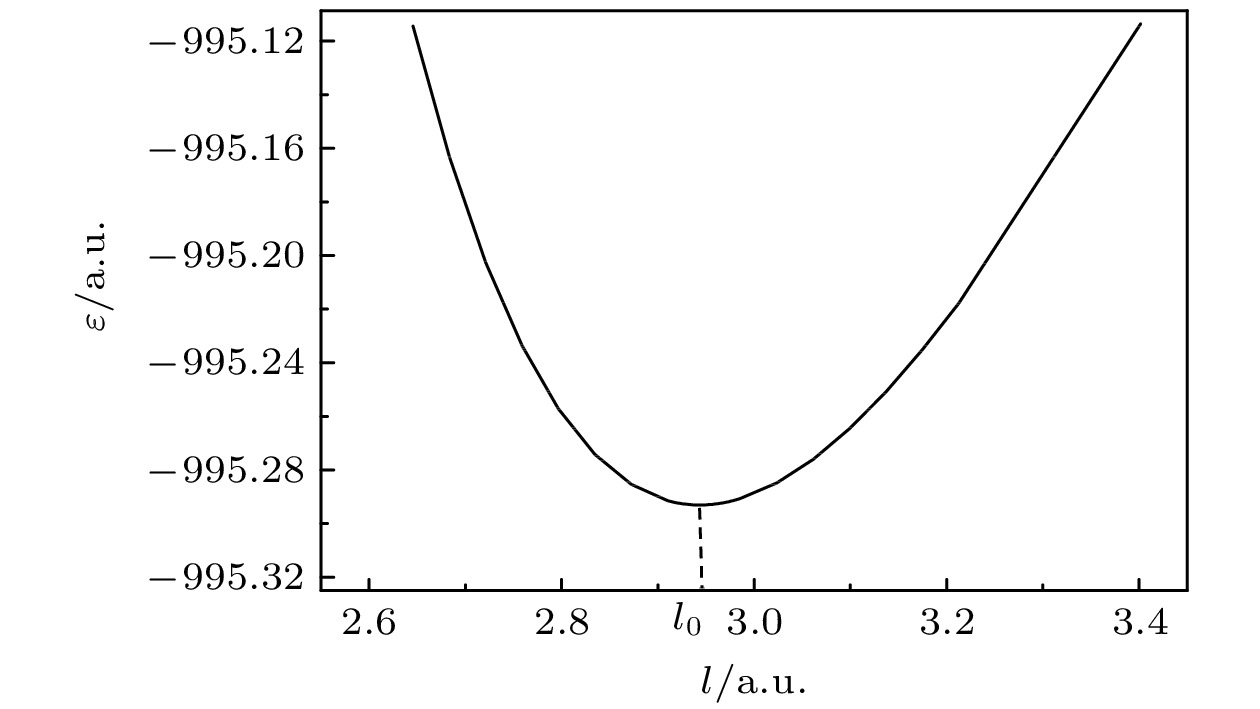
6molecule. It has 7 atoms and 70 electrons, hence there exists a large amount of electronic correlation energy to be calculated. The MRCI is an effective method to calculate the correlation energy. But by now no MRCI result of SF
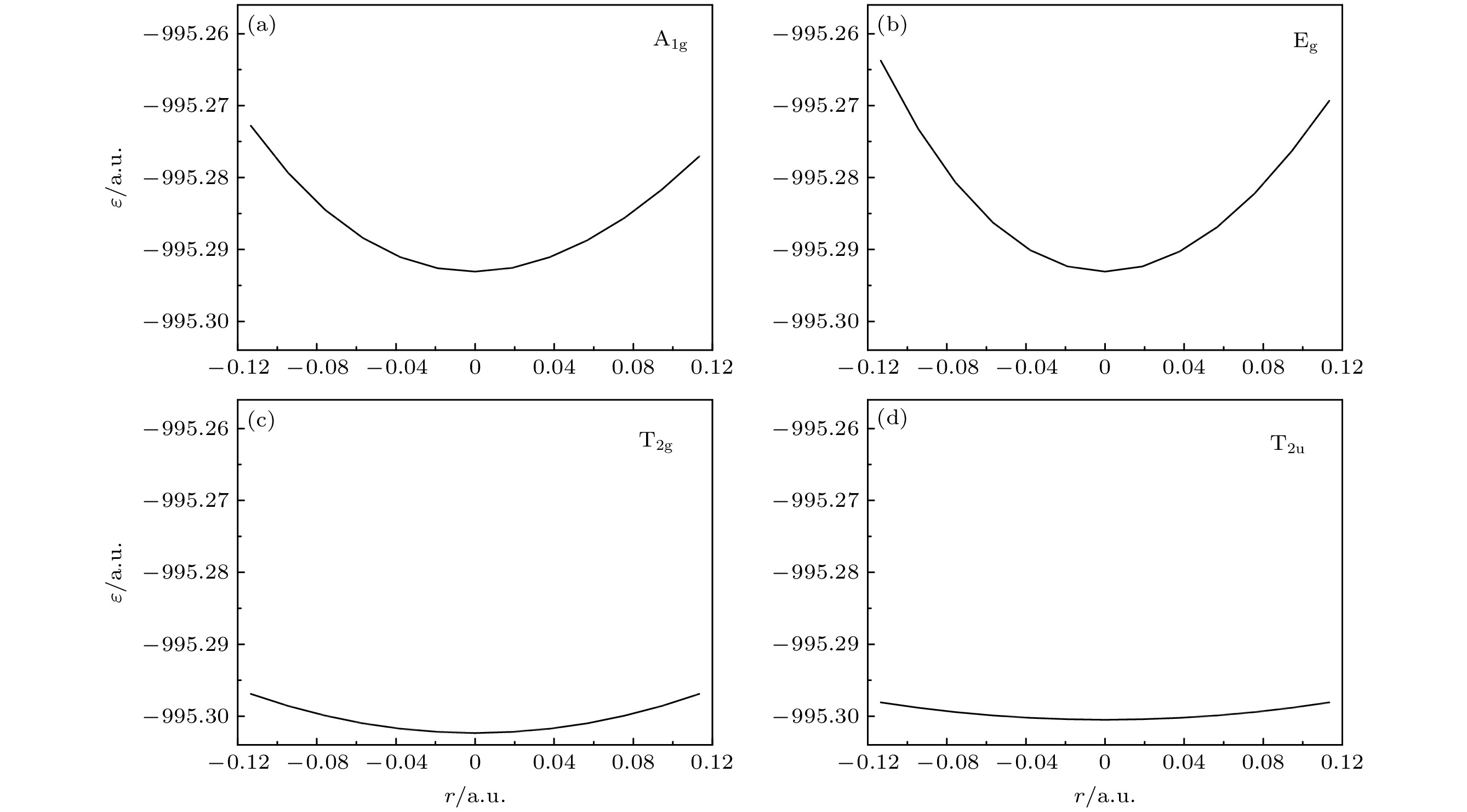
6vibrational frequencies has been reported. So here we use MRCI/6-311G* to calculate the potential curves of A
1g, E
g, T
2gand T
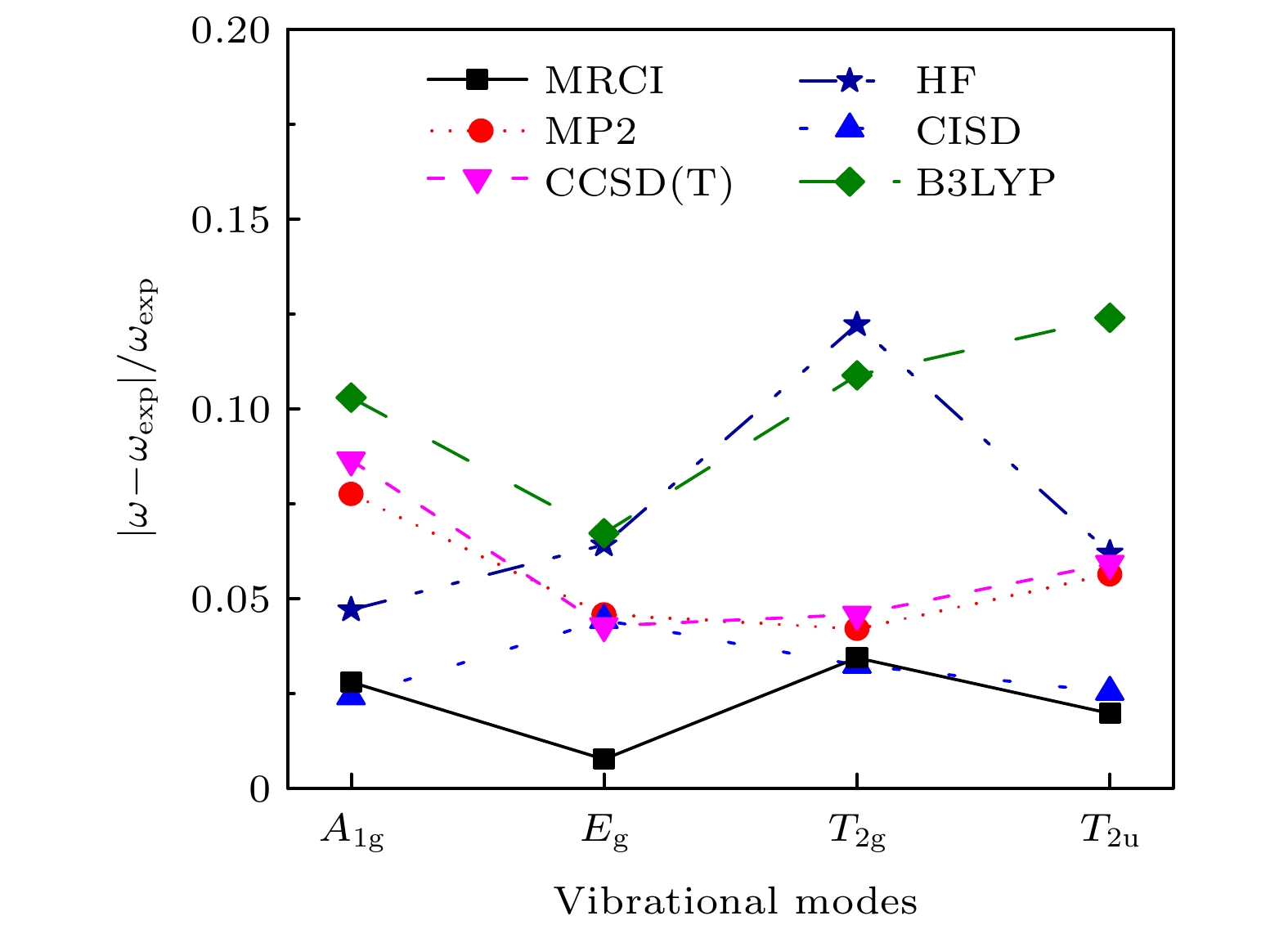
2uvibrational modes separately, deduce their expressions, then use the expressions to fit the curves, and finally obtain the vibrational frequencies. The results are then compared with those obtained by other theoretical methods including HF, MP2, CISD, CCSD(T) and B3LYP methods through using the same 6-311G* basis set. It is shown that the relative error to experimental result of the MRCI method is the least in the results from all these methods.












 DownLoad:
CSV
DownLoad:
CSV


 DownLoad:
DownLoad:




