There have been some theoretical studies of high pressure phase transition behavior of BaF
2, while in most cases the attention is paid mainly to the optical and electrical properties of BaF
2under increasing pressure. To date, there has been still a lack of theoretical explanation for the hysteresis phenomenon of high-pressure phase of BaF
2when the pressure is released. In addition, the pressure-dependent behavior of the BaF
2band gap is still under controversy, and there are few studies of its high-pressure Raman spectra. Therefore, first principle is used to make a supplementary calculation of the high pressure behavior of BaF
2. For a given pressure
Pand temperature
T, the thermodynamic stable phase has the lowest Gibbs free energy. The calculations are performed at zero temperature and hence, the Gibbs free energy becomes equal to the enthalpy. Thus, the variation of enthalpy is calculated as a function of pressure to study the high-pressure phase stability of BaF
2based on density functional theory as implemented in the Vienna ab initio simulation package (VASP). The results show that the BaF
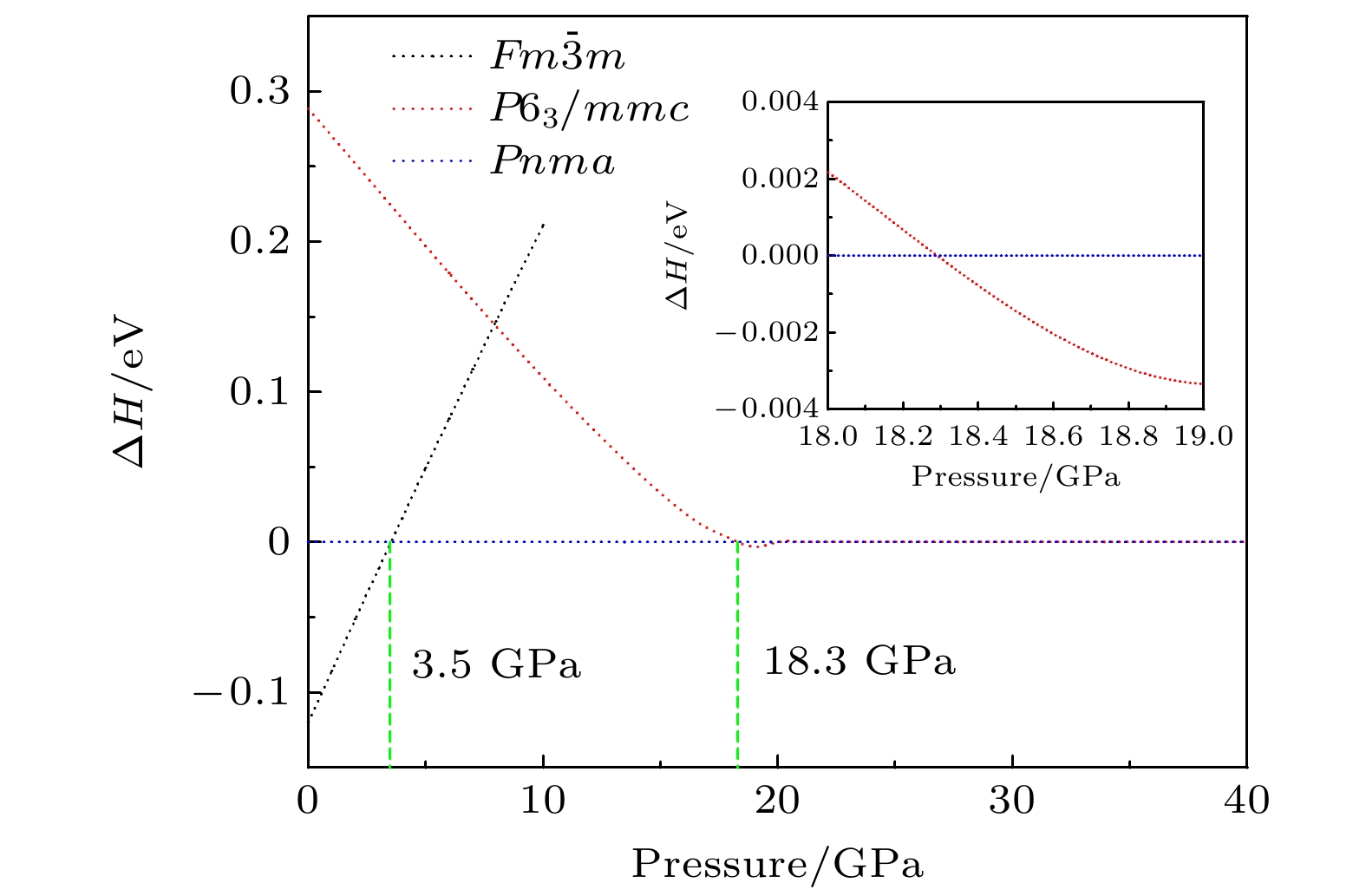
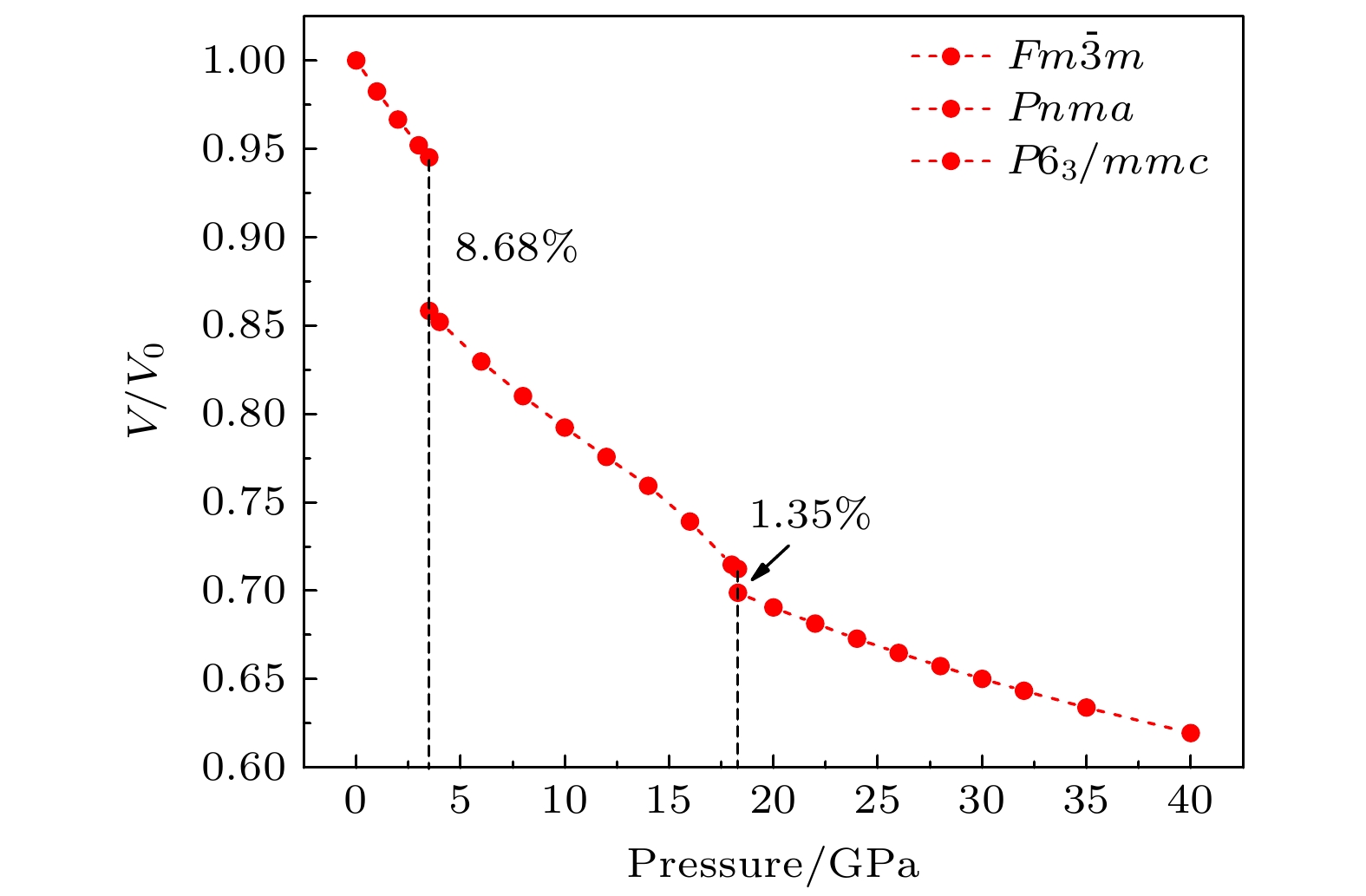
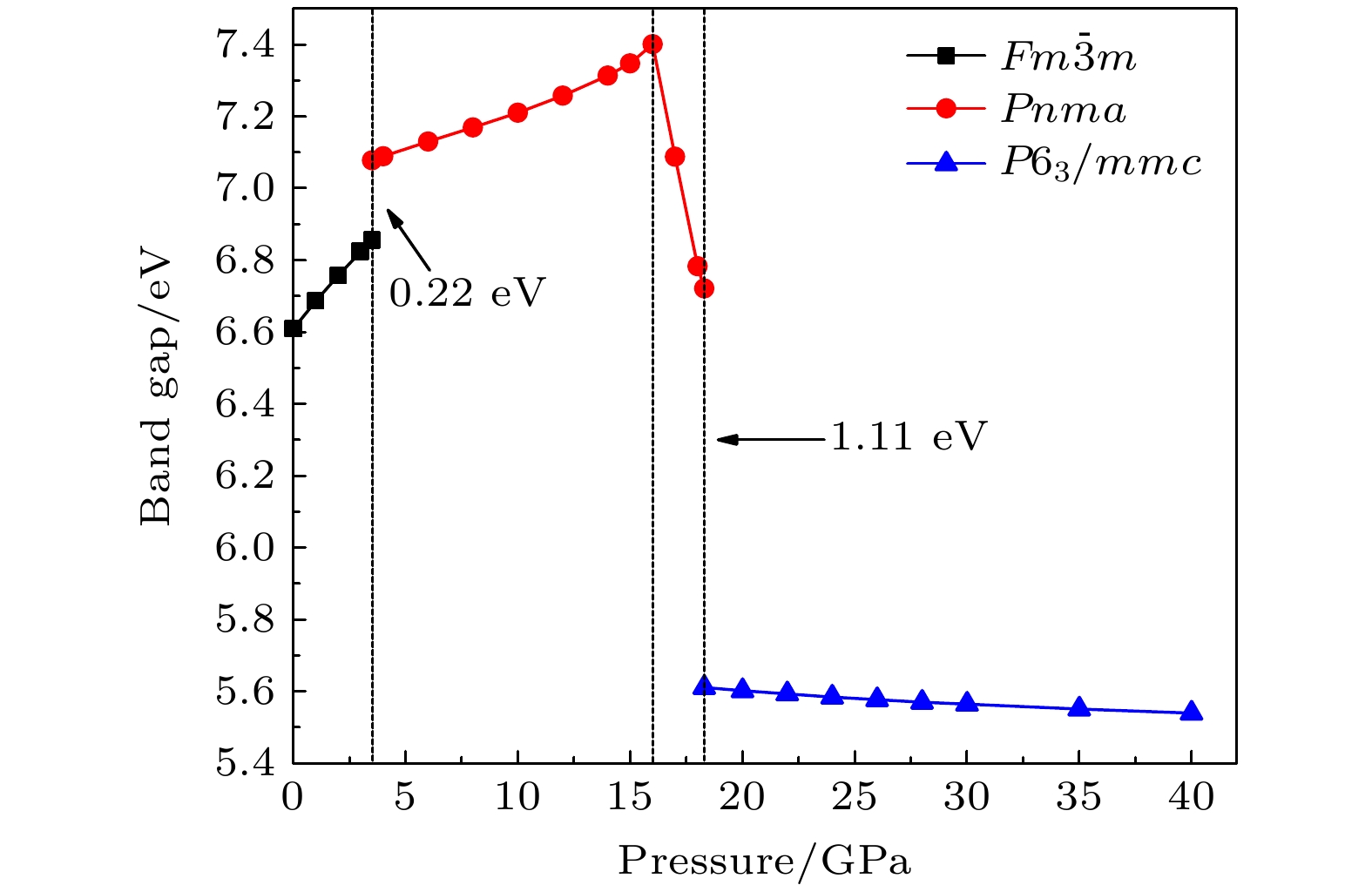
2undergoes two structural phase transitions from
Fm3
m(cubic) to
Pnma(orthorhombic) and then to
P6
3/
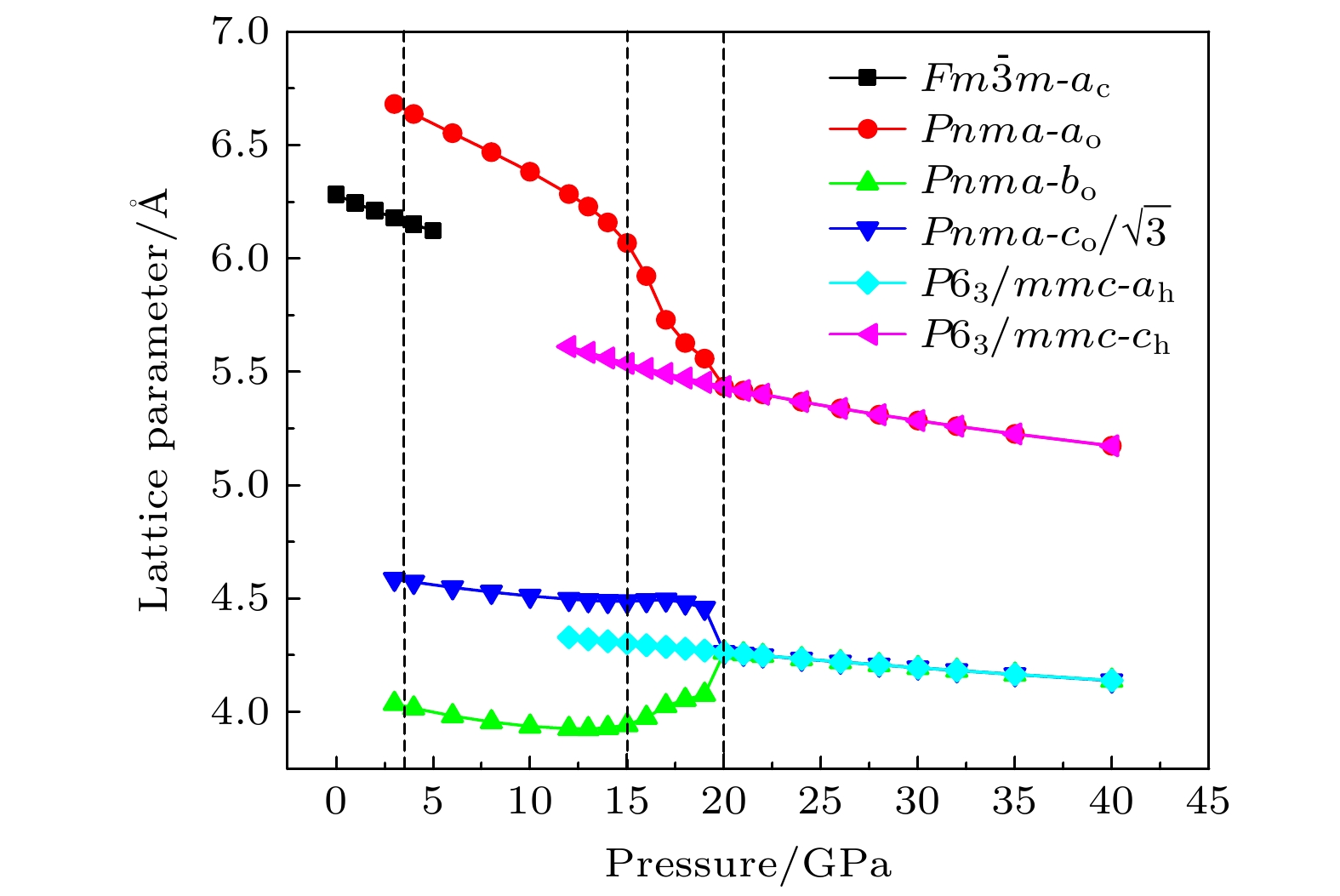
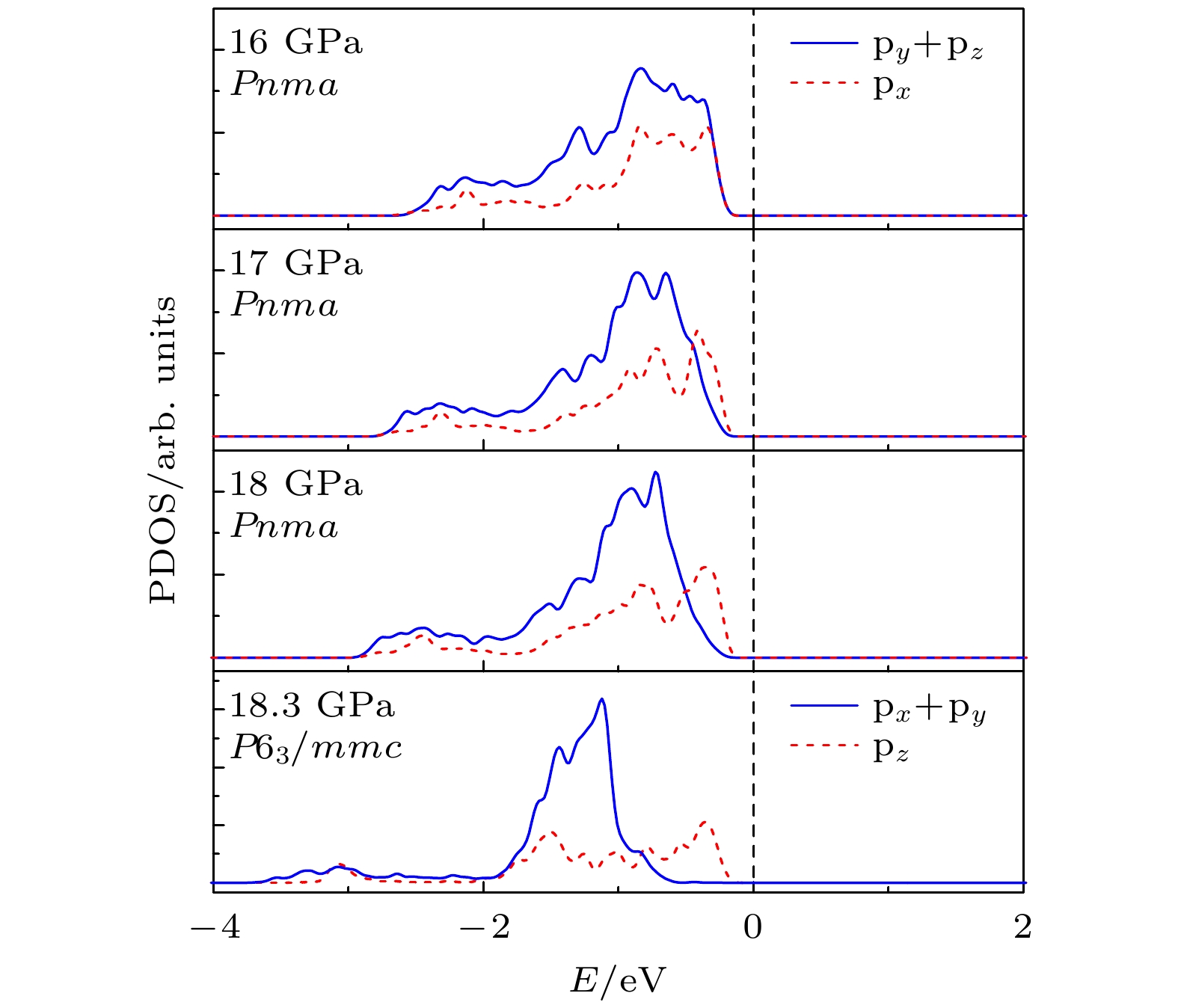
mmc(hexagonal) with increasing pressure, and their corresponding transition pressures are 3.5 and 18.3 GPa, respectively. By calculating the evolution of lattice constant with pressure, it is found that at about 15 GPa (near the second phase transition pressure), the lattice constants of the
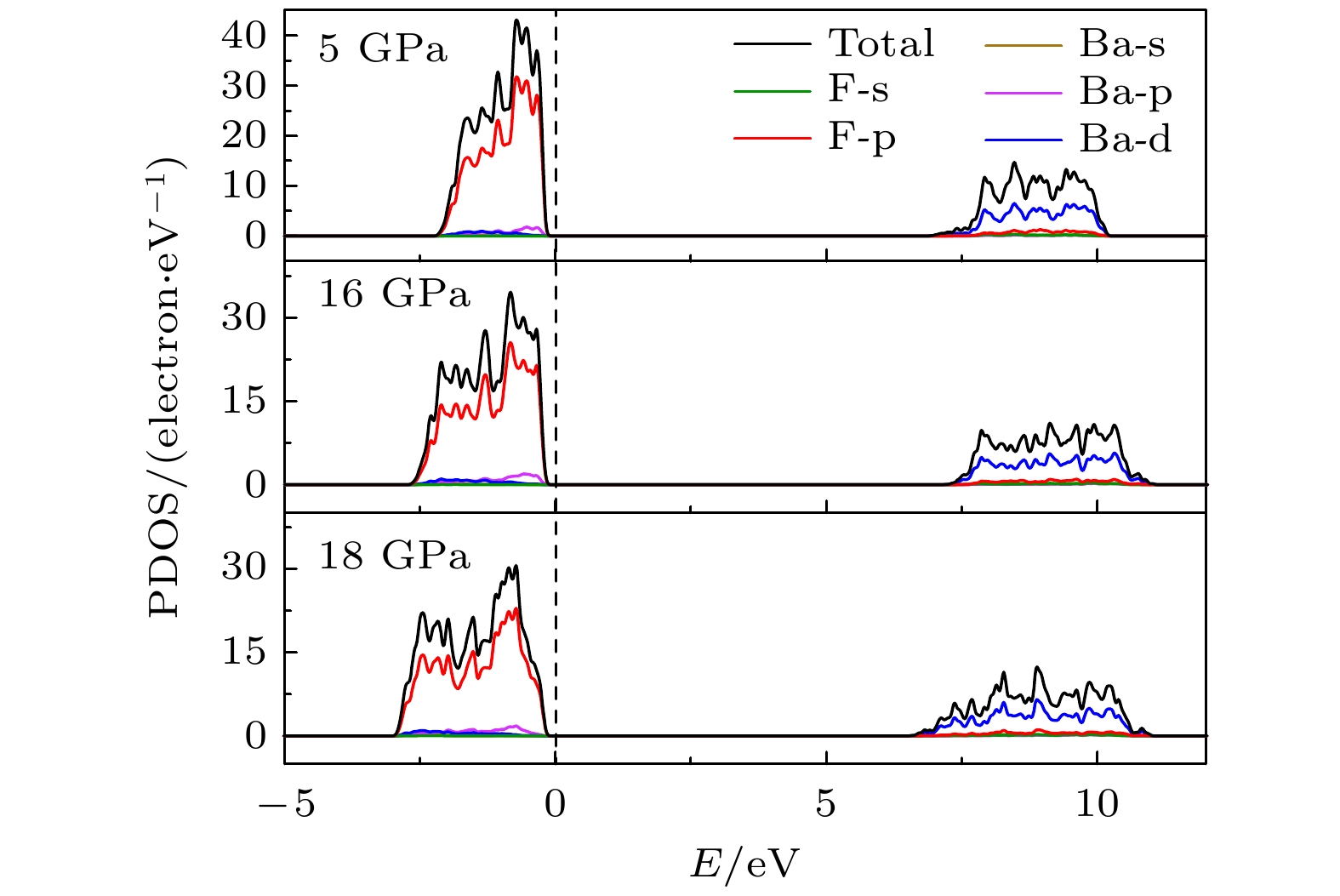
Pnmastructure show abnormal behavior (a slight increase in
b
oand a slight decrease in
a
o). We suggest that this behavior leads the band gap to decrease, indicated by analyzing the calculated results of
Pnmastructure of other materials. The
Pnmastructure completely transforms into
P6
3/
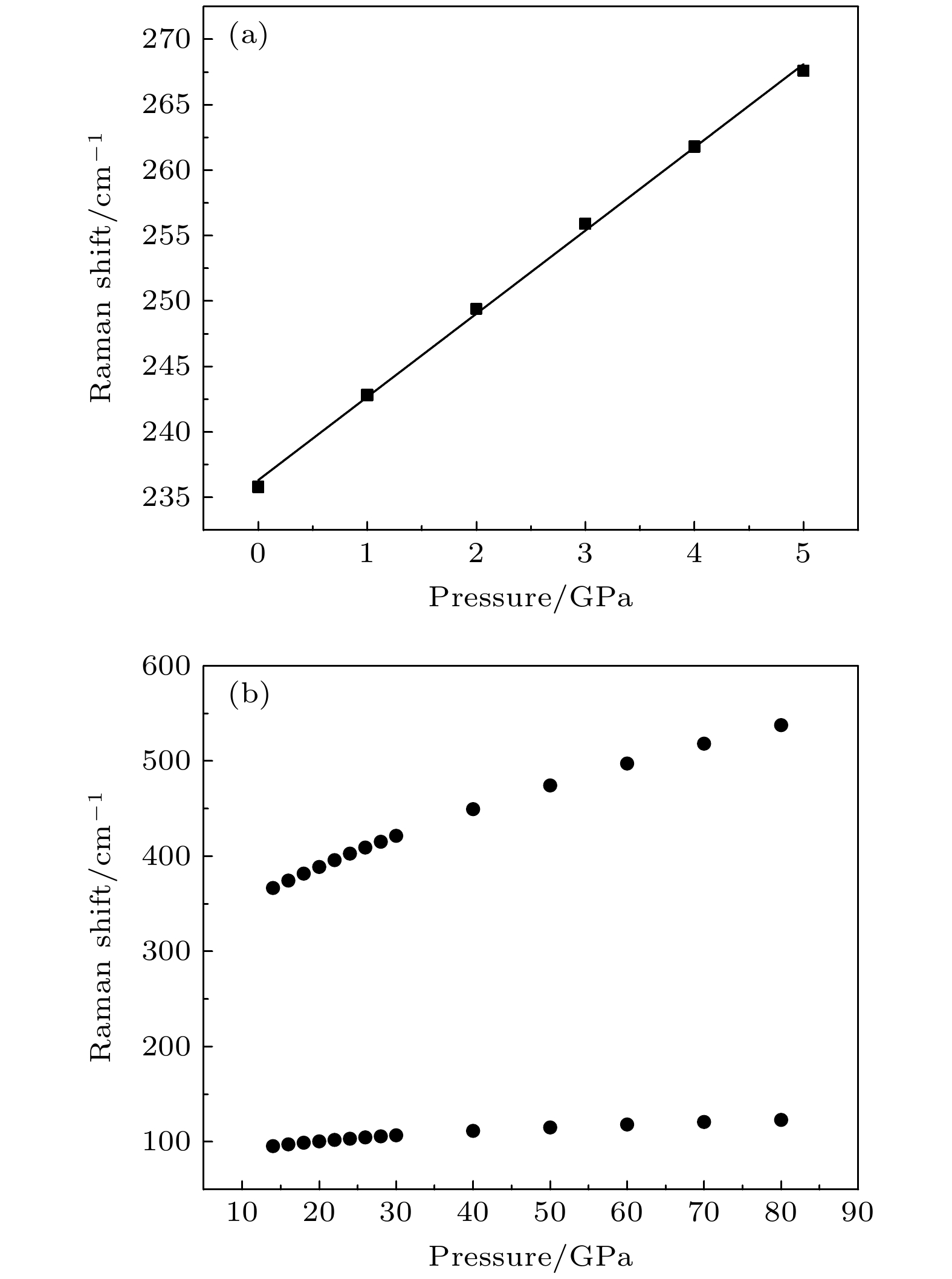
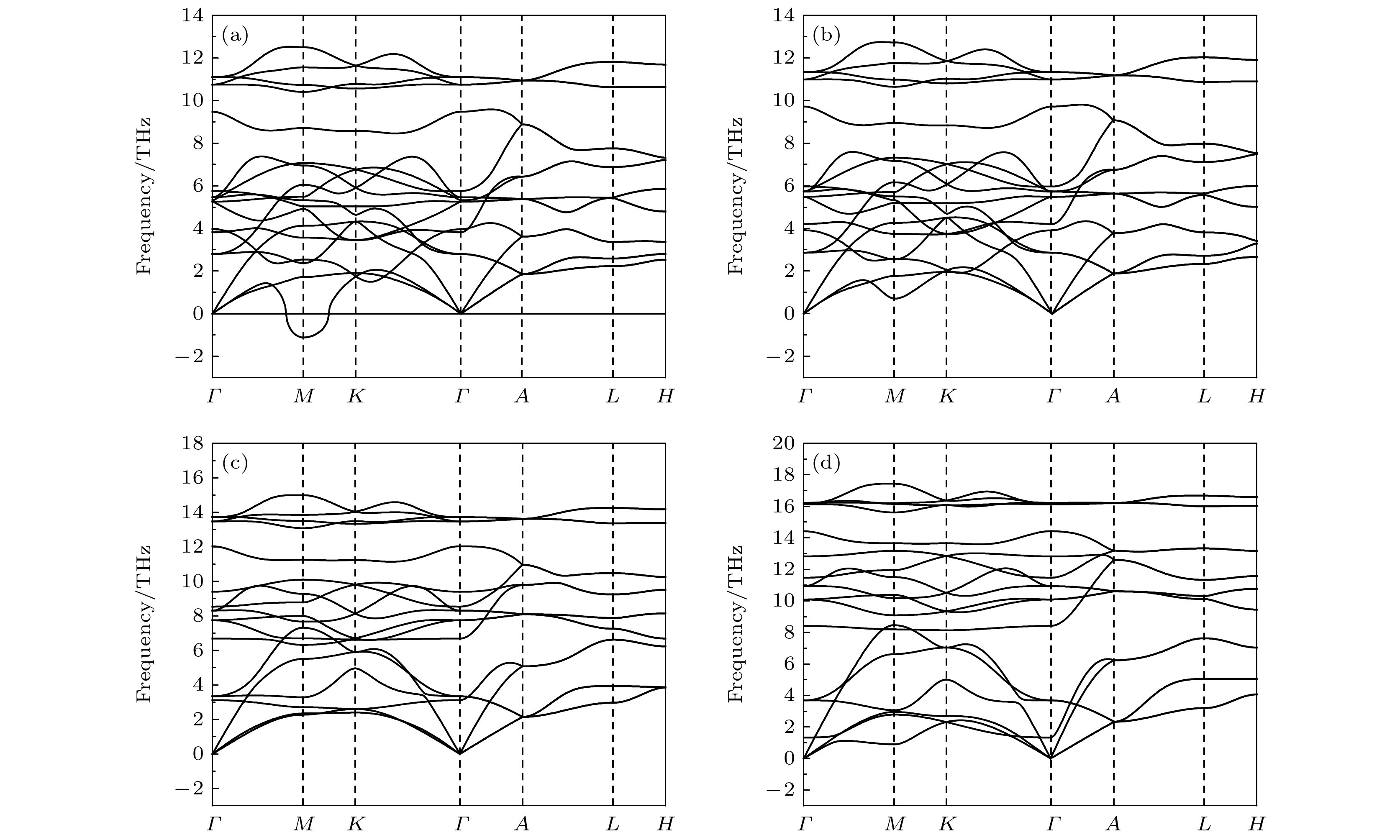
mmcstructure at about 20 GPa. By analyzing the phonon dispersion curves of BaF
2as a function of pressure, the structural stability information of the material can also be obtained. Then the density functional perturbation theory (DFPT) is used to calculate the phonon dispersion curves of BaF
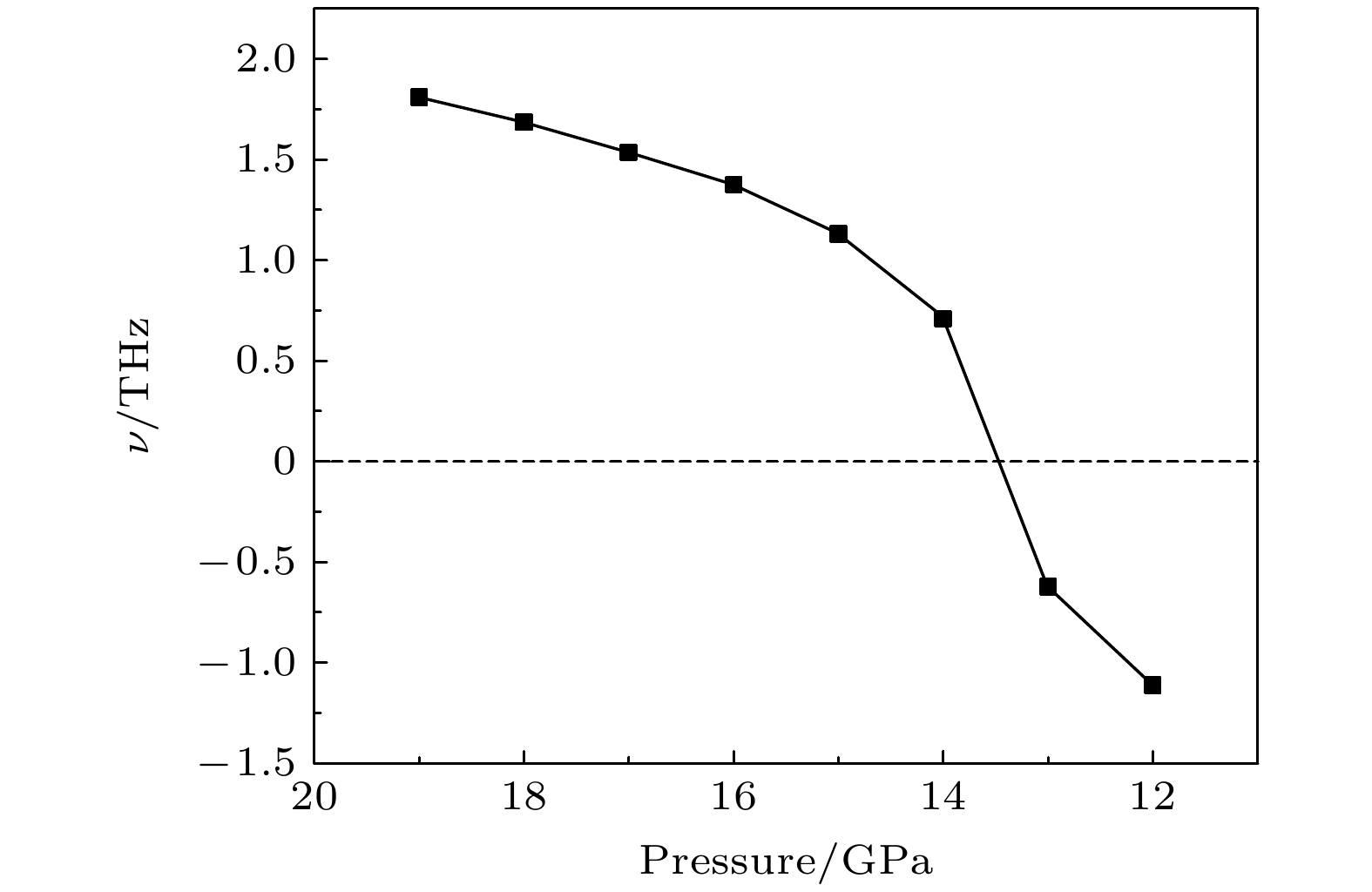
2by VASP code and Phonopy code. The hysteresis phenomenon of the
P6
3/
mmcstructure, when the pressure is released, is explained by the kinetic stability. The results predict that the
P6
3/
mmcstructure can be stabilized at least to 80 GPa.












 DownLoad:
CSV
DownLoad:
CSV


 DownLoad:
DownLoad:







