Lithium-rich manganese-based ternary cathode material for lithium-ion batteries, Li
1.208Ni
0.333Co
0.042Mn
0.417O
2, has excellent structural stability and electrochemical stability due to its high Ni content. In order to understand the physical properties of this lithium-rich material, its crystal structure, electronic structure and defect properties are calculated by employing the first-principles method based on the density functional theory. The obtained electronic structure shows that Li
1.208Ni
0.333Co
0.042Mn
0.417O
2is a magnetic semiconductor with a direct band gap of 0.47 eV. The analysis of the electronic state suggests that the electronic state at the valence band maximum (VBM) is the hybridization of p
x, p
y, p
zorbitals of oxygen and the d
xy, d
yz, d
xzorbitals of Ni-atom. The electronic state at the conduction band minimum has similar characteristics to those at the VBM, however, part of Ni-
${3{\rm{d}}}_{{x}^{2}-{y}^{2}}$

and Mn-
${3{\rm{d}}}_{{x}^{2}-{y}^{2}}$
, and Mn-
${3{\rm{d}}}_{yz}$

also contribute to the electronic hybridizations. The charge density difference calculations indicate that the bonding between O and transition metal atoms are through the mixture of covalent bond with ionic bond. The vacancy formation of a single metal atom is also calculated. The results show that the volumes of the defect systems containing metal vacancies are all reduced in comparison with the volume of perfect lattice. The volume change is the largest for the formation of Mn-vacancy, while the volume is almost unchanged with Co atoms extracted. The vacancy formation energies of the metals are
E
f(Mn) >
E
f(Co) >
E
f(Ni), and the vacancy formation energy of Mn is significantly higher than those of Ni and Co, indicating that the presence of Mn provides a major structural stability for the material. The calculated charge density differences also show that the formation of metal vacancies influences only the charge distribution of the oxygen atoms around the vacancy, showing the local character of the vacancy effect. Since the formation of metal vacancy breaks the bonding between the metal and the surrounding oxygen atoms, the O-2p states near the Fermi surface are significantly increased as shown in the calculated electronic density of states. Such a picture suggests that the electrons on oxygen atoms in vicinity of the metal vacancies become freer.












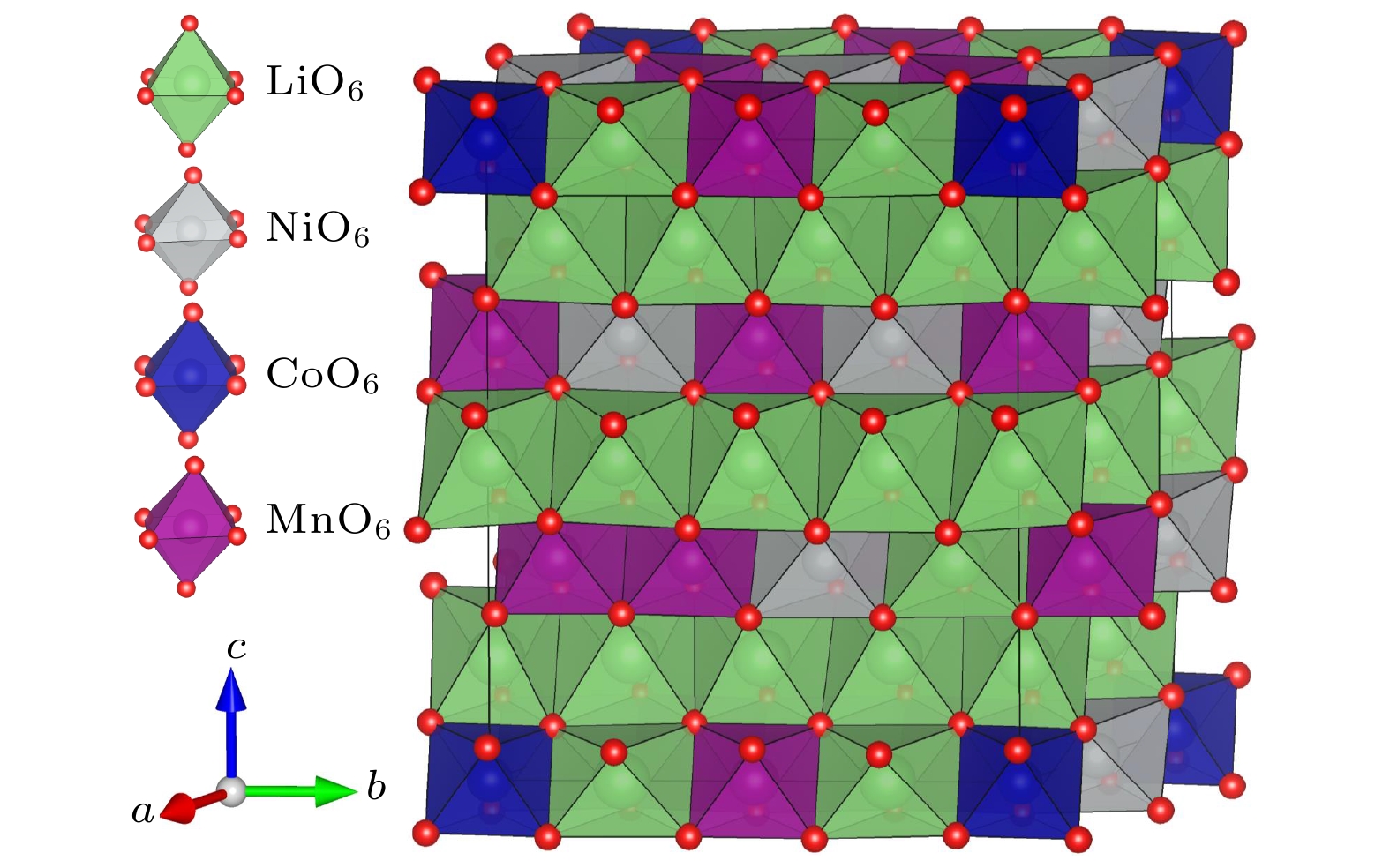
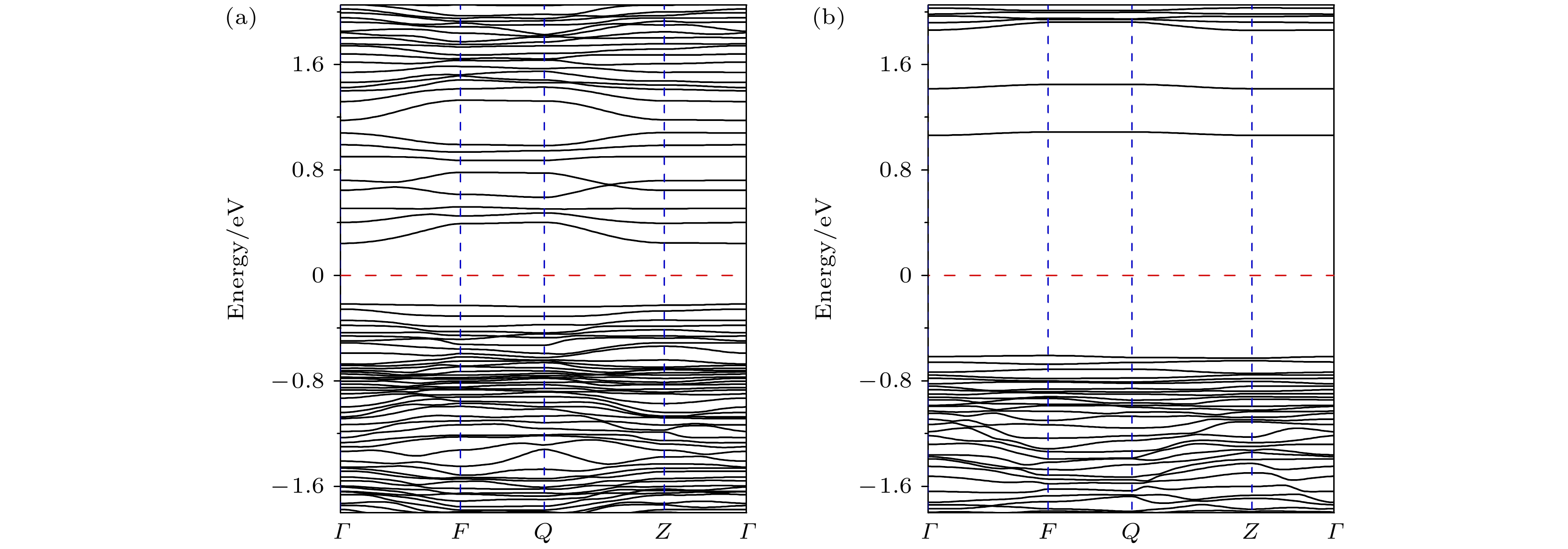
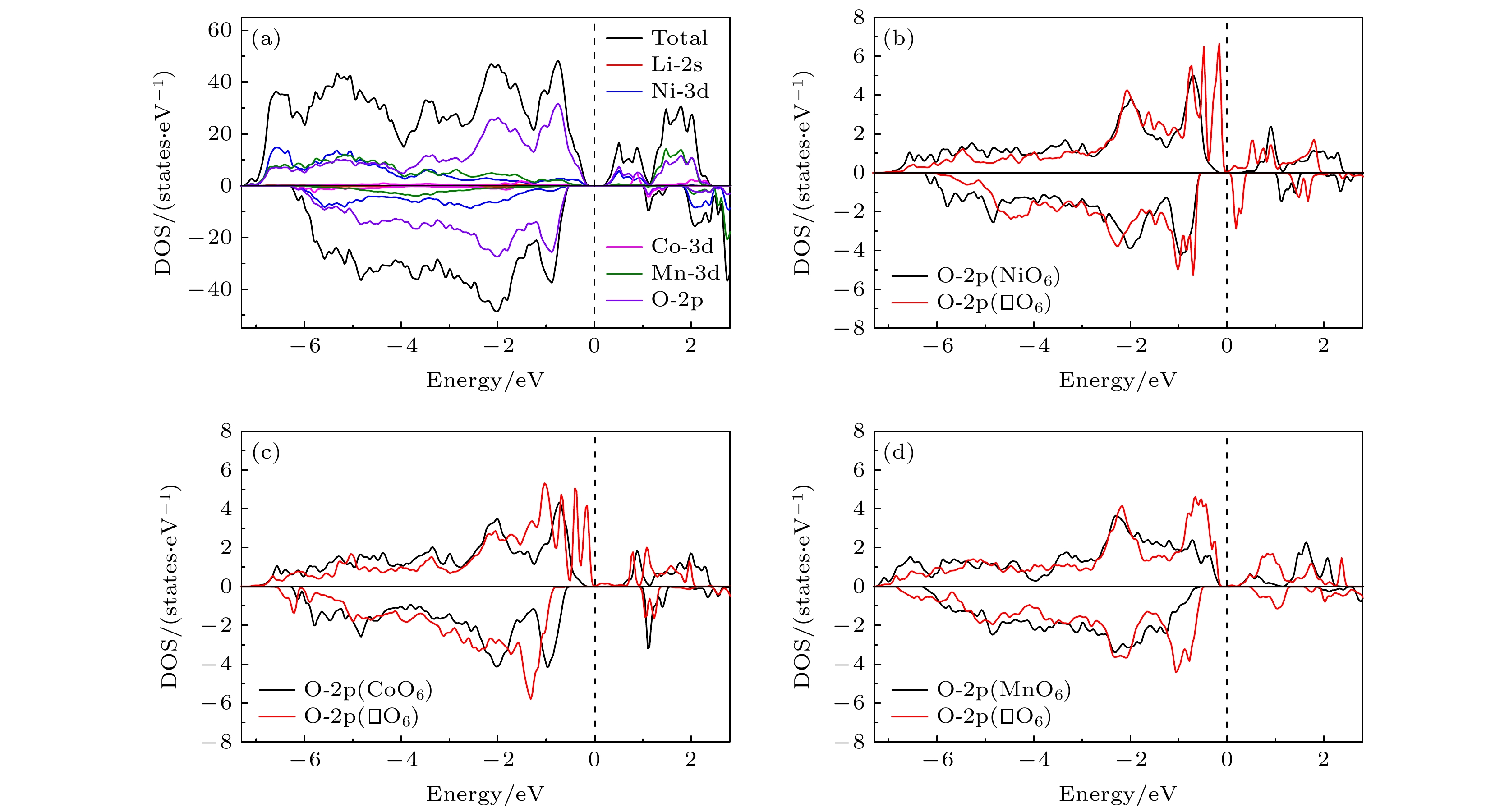
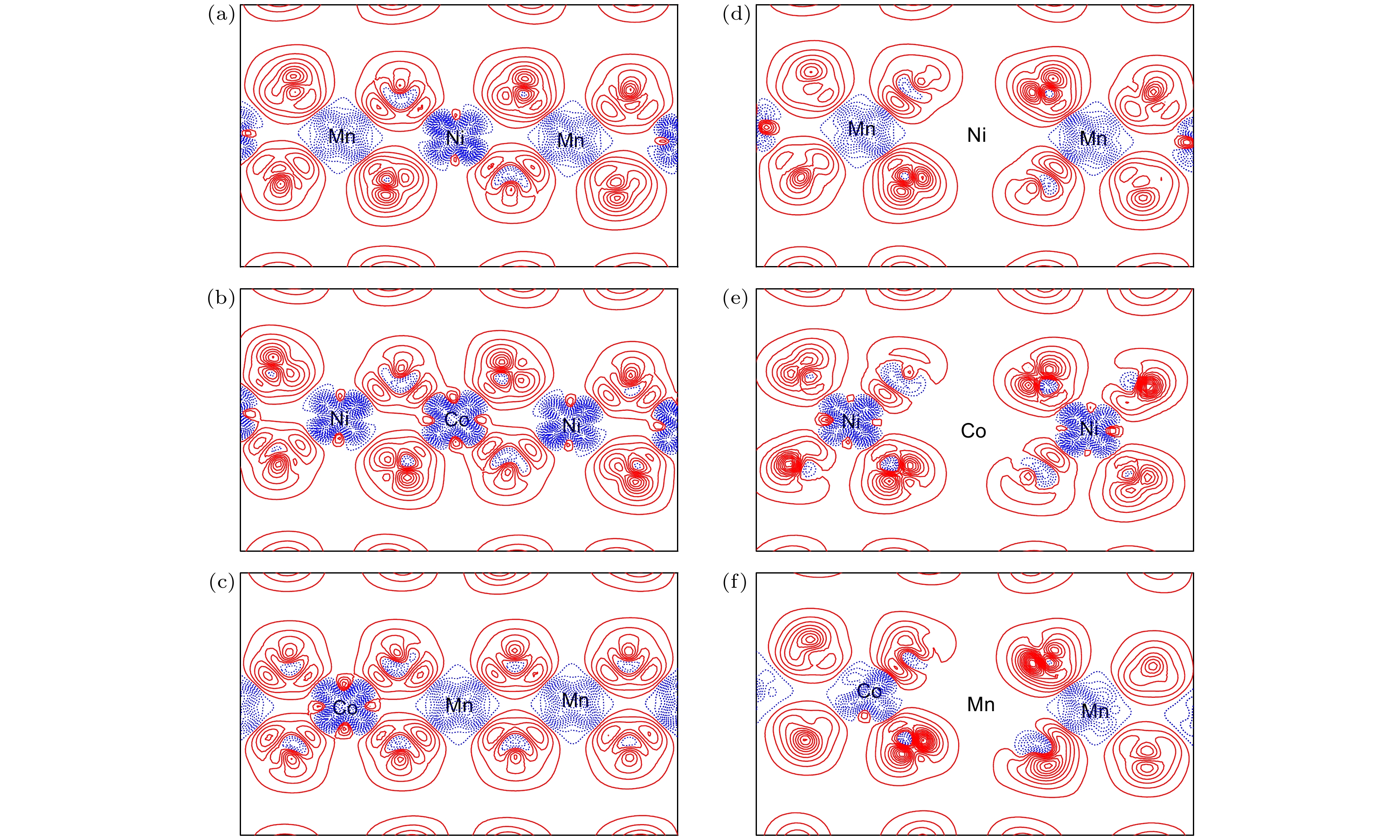
 DownLoad:
CSV
DownLoad:
CSV




 DownLoad:
DownLoad:


