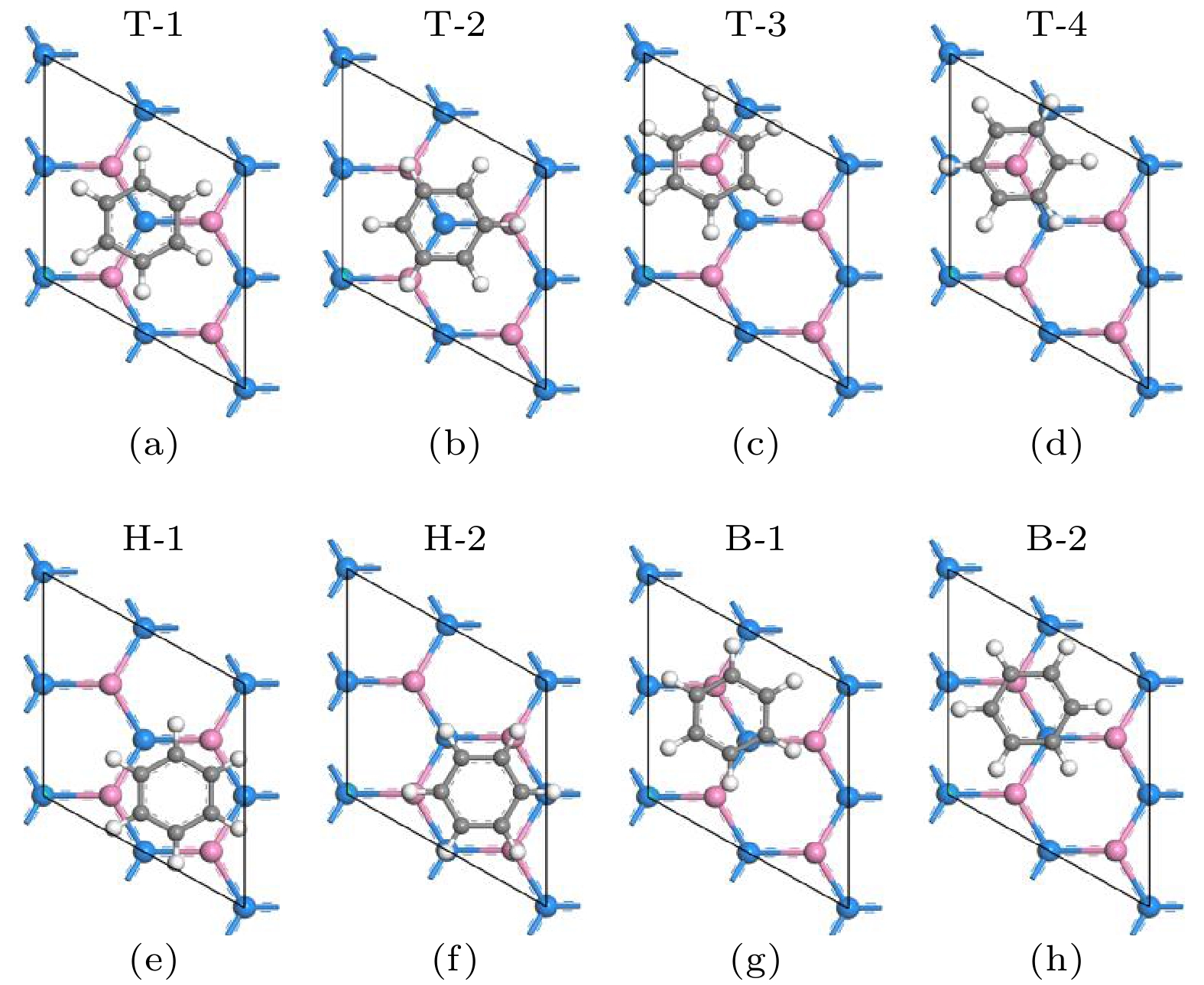
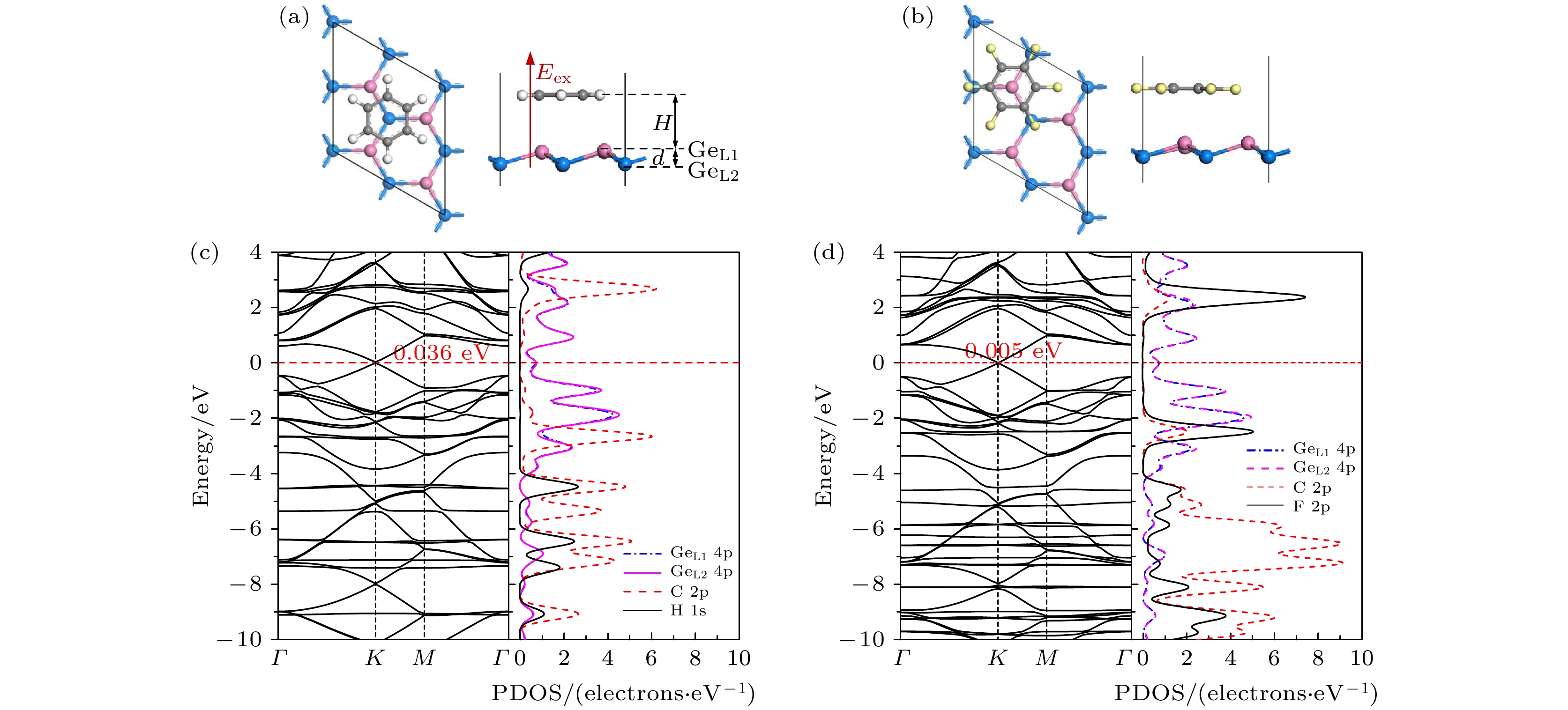
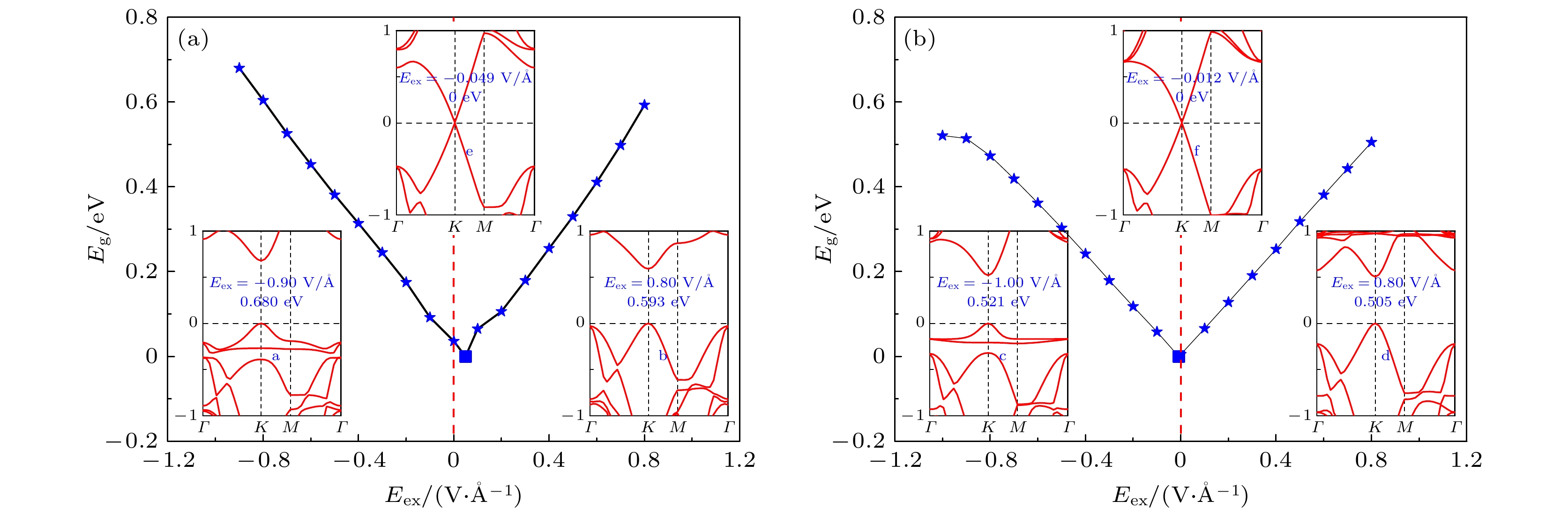
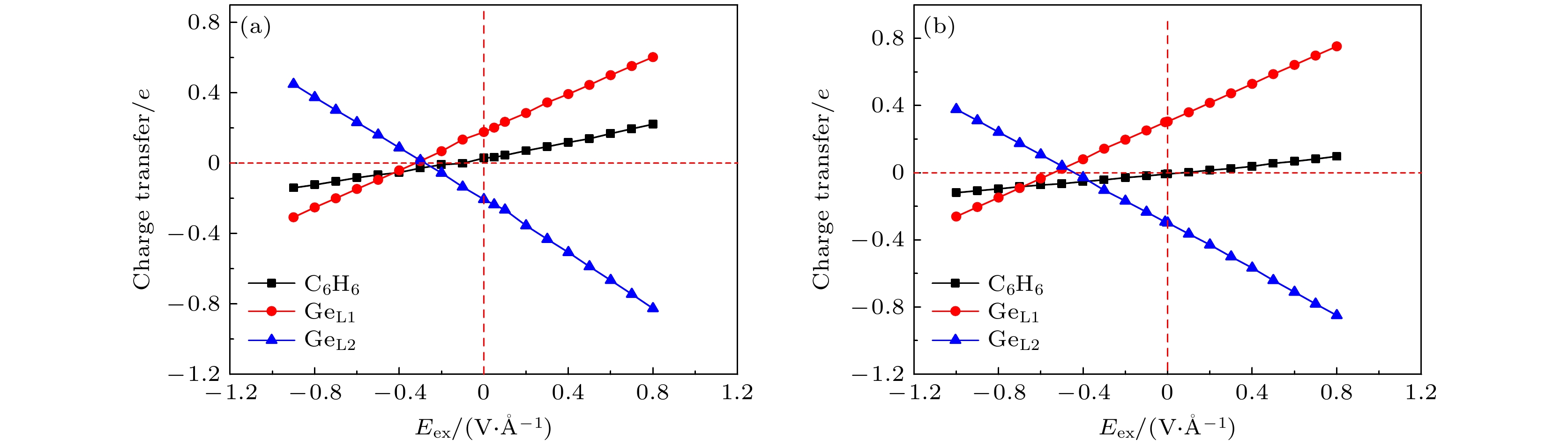
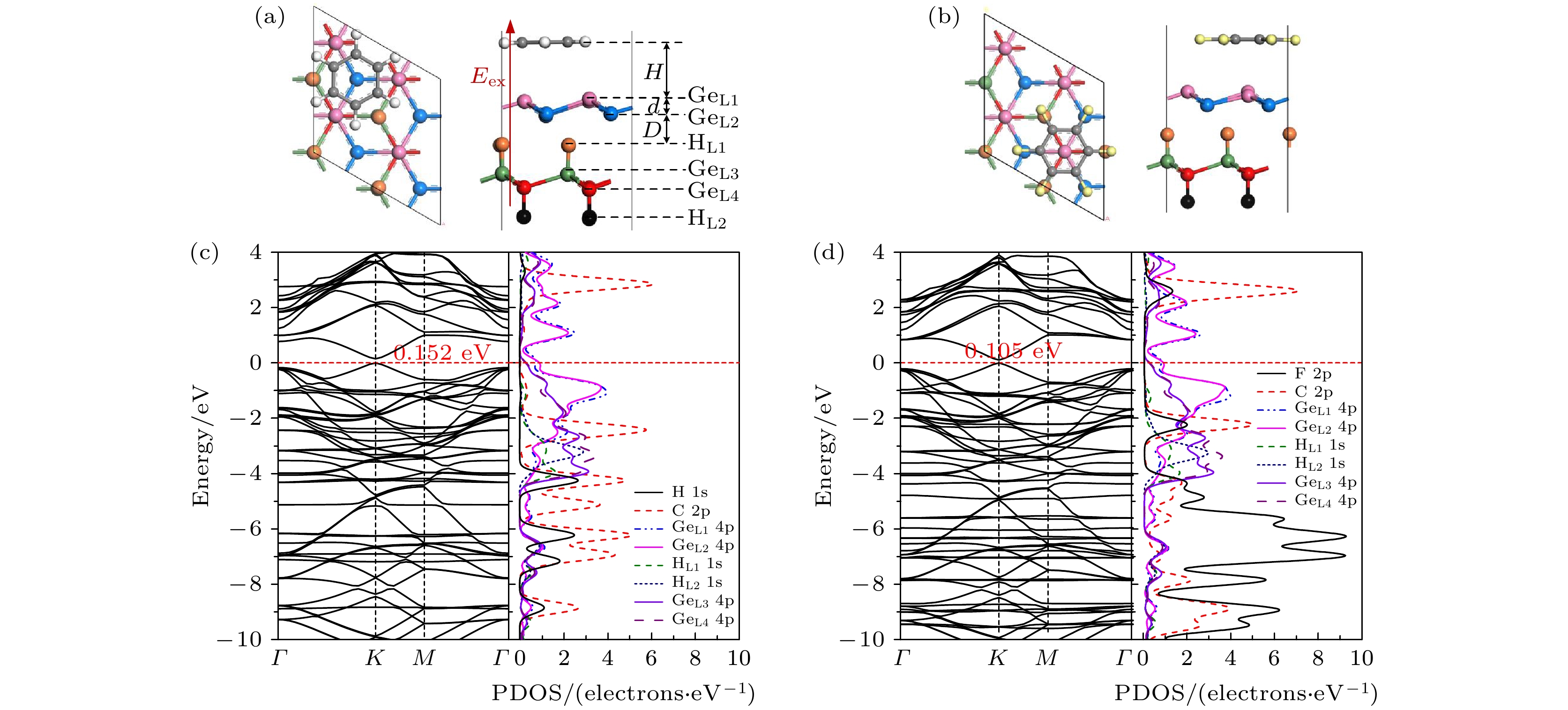
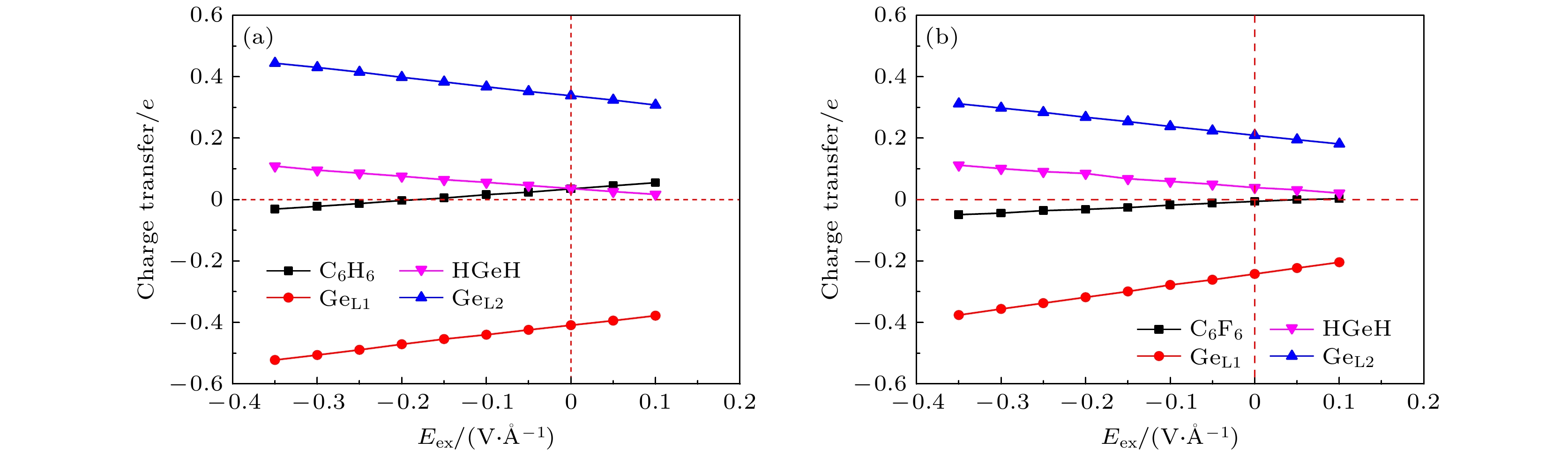
The development potential of germanene-based integrated electronics originates from its high carrier mobility and compatibility with the existing silicon-based and germanium-based semiconductor industry. However, the small band gap energy band (Dirac point) of germanene greatly impedes its application. Thus, it is necessary to open a sizeable band gap without reducing the carrier mobility for the application in logic circuits. In this study, the effects of organic molecule (benzene or hexafluorobenzene) adsorption and substrate on the atomic structures and electronic properties of germanene under an external electric field are investigated by using density functional theory calculations with van der Waals correction. For benzene/germanene and hexafluorobenzene/germanene systems, four different adsorption sites are considered, with the center of the organic molecules lying directly atop the upper or lower Ge atoms of germanene, in the Ge-Ge bridge center, and on the central hollow ring. Meanwhile, different molecular orientations at each adsorption site are also considered. Thus, there are eight high-symmetry adsorption configurations of the systems, respectively. According to the adsorption energy, we can determine the most stable atomic structures of the above systems. The results show that the organic molecule adsorption can induce the larger buckling height in germanene. Both the adsorption energy and interlayer distance indicate that there is no chemical bond between the organic molecules and germanene. Mulliken population analysis shows that a charge redistribution in the two sublattices in germanene exists since benzene is an electron donor molecule and hexafluorobenzene is an electron acceptor molecule. As a result, the benzene/germanene system exhibits a relatively large band gap (0.036 eV), while hexafluorobenzene/germanene system displays a small band gap (0.005 eV). Under external electric field, germanene with organic molecule adsorption can exhibit a wide range of linear tunable band gaps, which is merely determined by the strength of electric field regardless of its direction. The charge transfer among organic molecules and two sublattices in germanene gradually rises with the increasing the strength of electric field, resulting in the electron density around the sublattices in germanene unequally distributed. Thus, according to the tight-binding model, a larger band gap at the
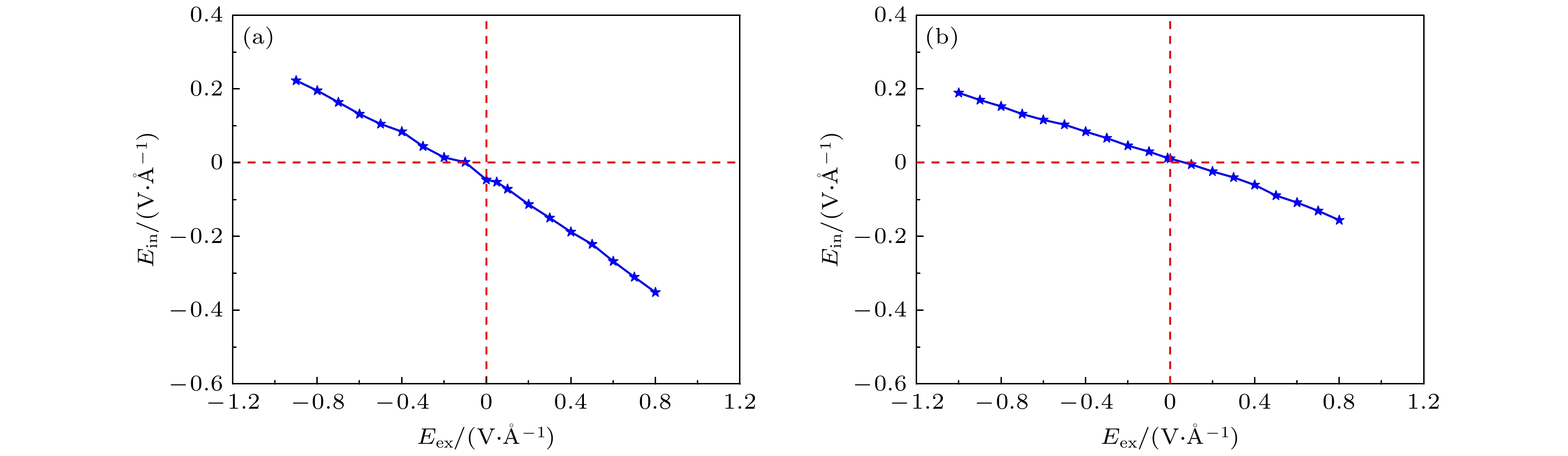
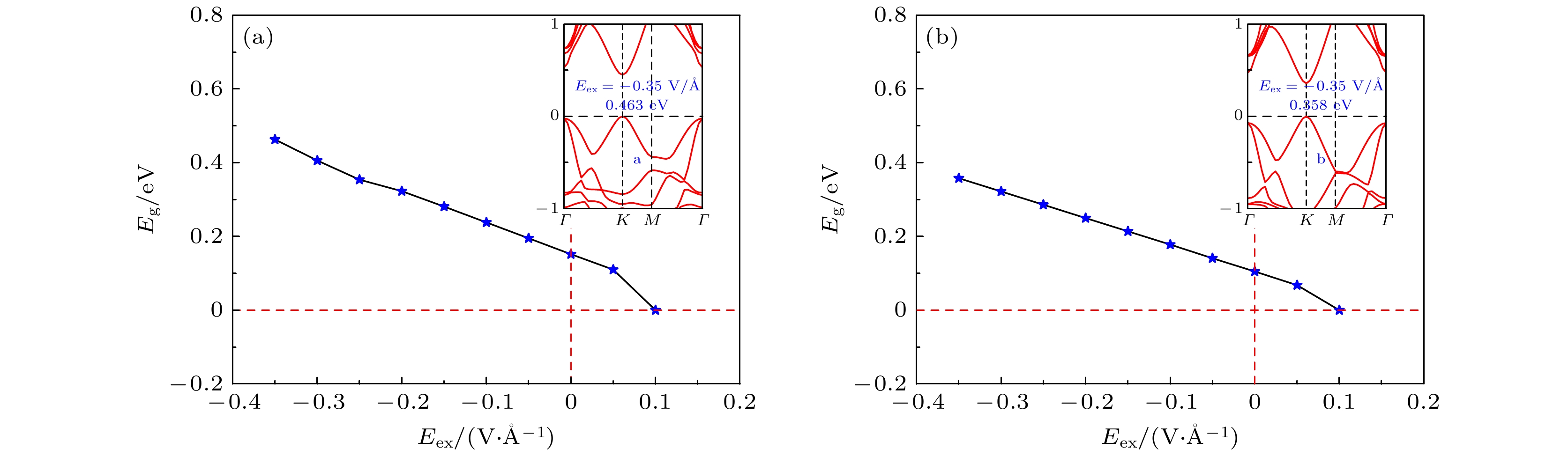
K-point is opened. When germanane (fully hydrogenated germanene HGeH) substrate is applied, the band gaps further widen, where the band gap of benzene/ germanene/germanane system can increase to 0.152 eV, and that of hexafluorobenzene/germanene/germanane system can reach 0.105 eV. The sizable band gap in germanene is created due to the symmetry of two sublattices in germanene destroyed by the dual effects of organic molecule adsorption and substrate. Note that both of organic molecules and substrate are found to non-covalently functionalize the germanene. As the strength of the negative electric field increases, the band gaps can be further modulated effectively. Surprisingly, the band gaps of the above systems can be closed, and reopened under a critical electric field. These features are attributed to the build-in electric field due to the interlayer charge transfer of the systems, which breaks the equivalence between the two sublattices of germanene. More importantly, the high carrier mobility in germanene is still retained to a large extent. These results provide effective and reversible routes to engineering the band gap of germanene for the applications of germanene to field-effect transistor and other nanoelectronic devices.












 DownLoad:
CSV
DownLoad:
CSV


 DownLoad:
DownLoad:






