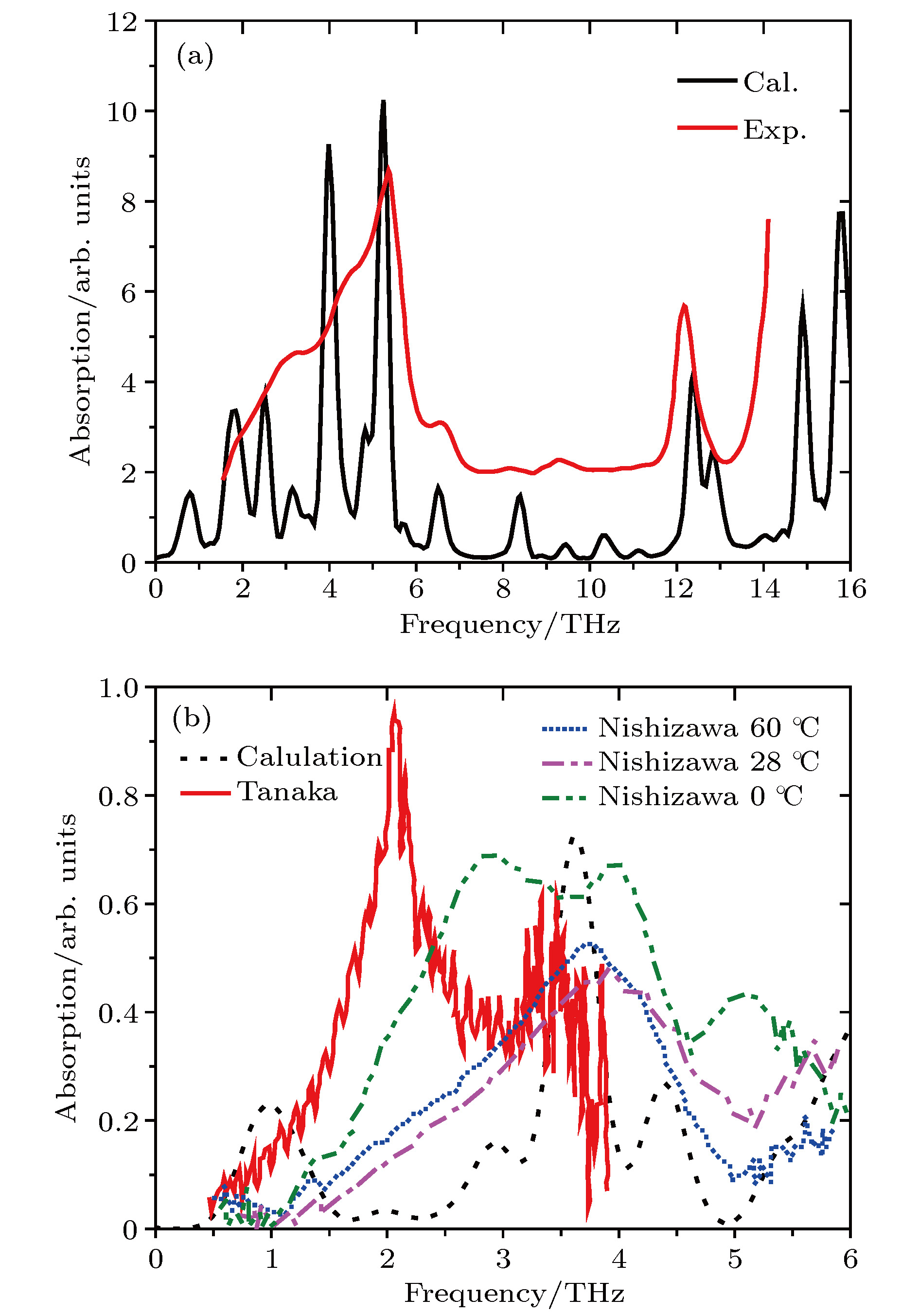
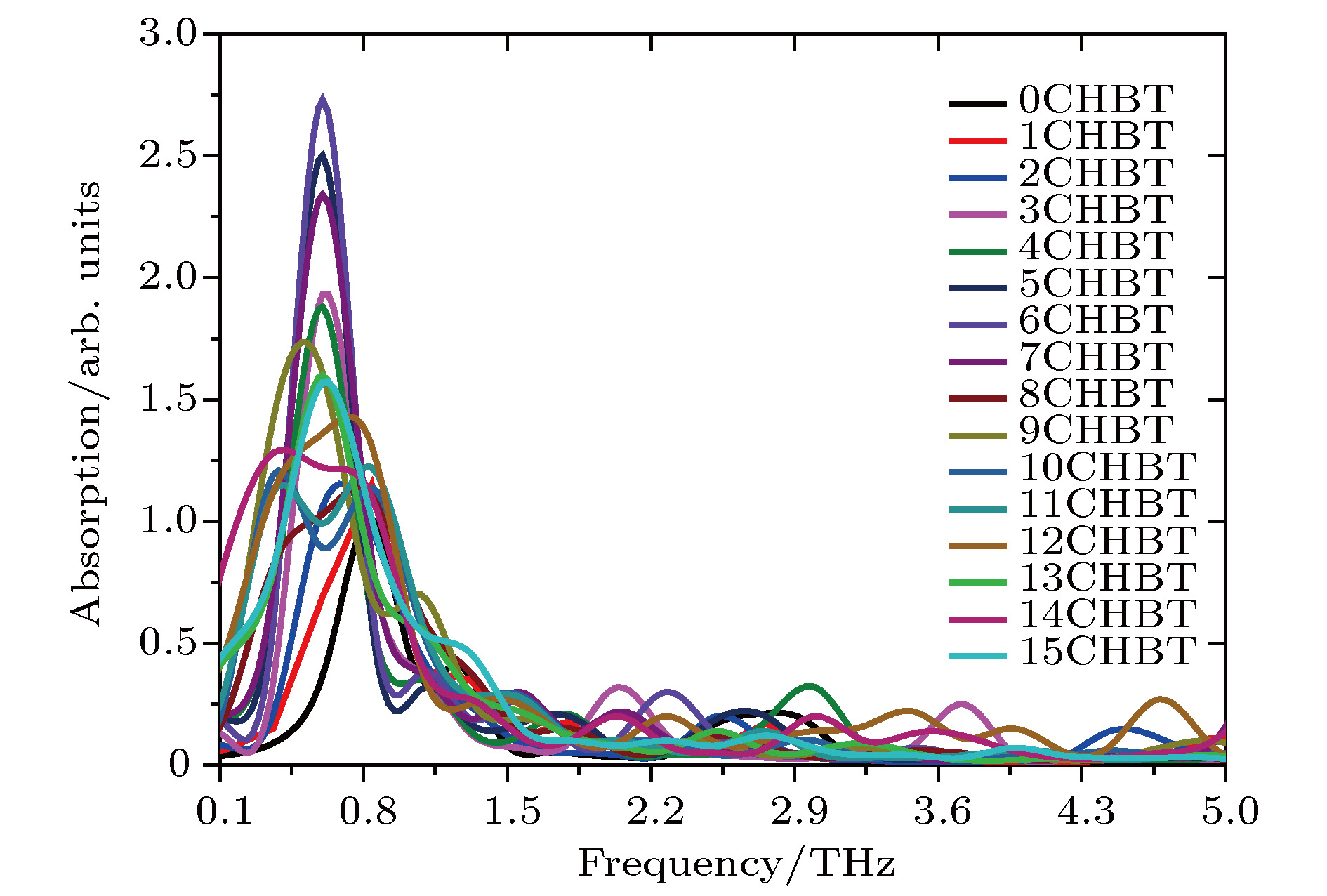
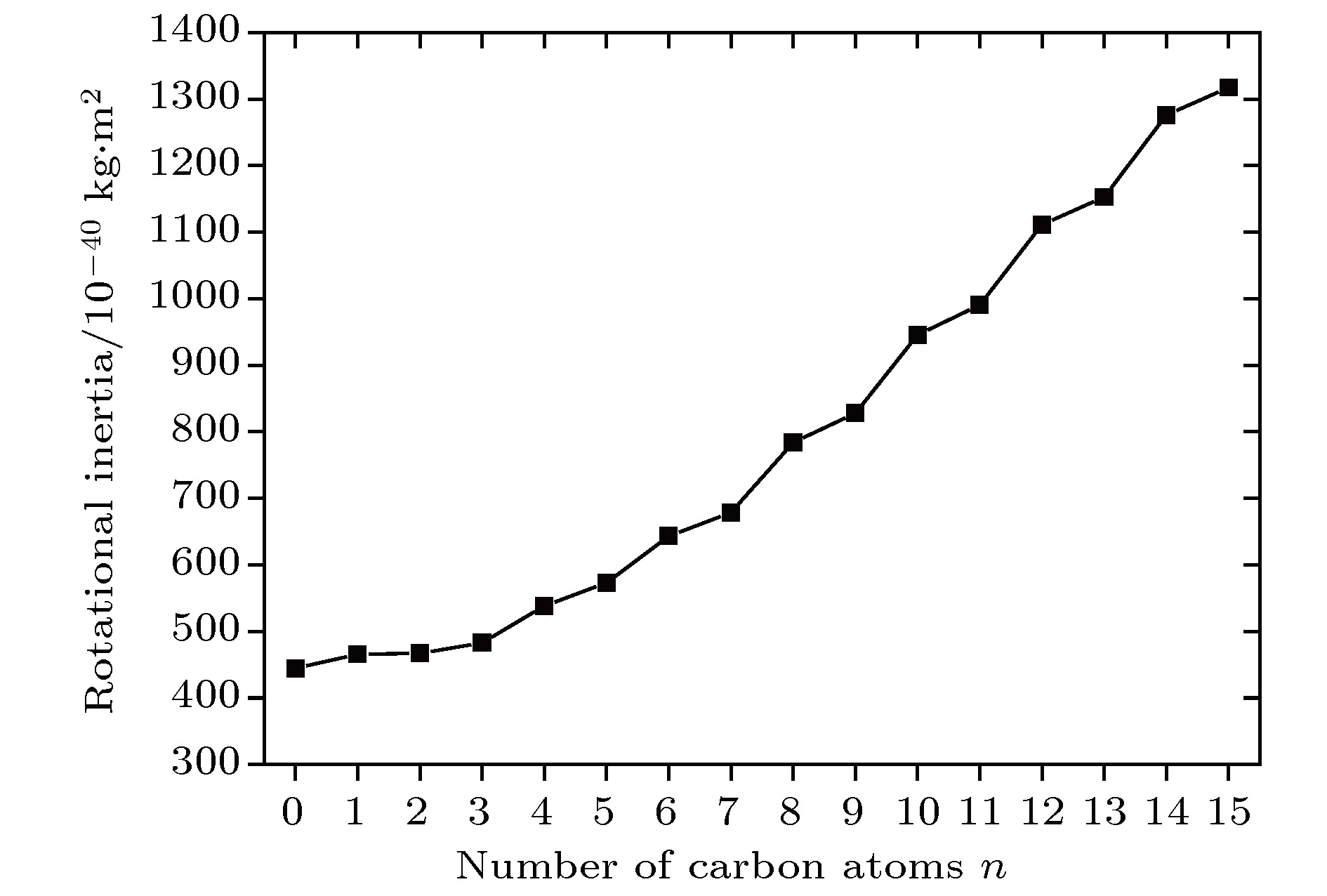
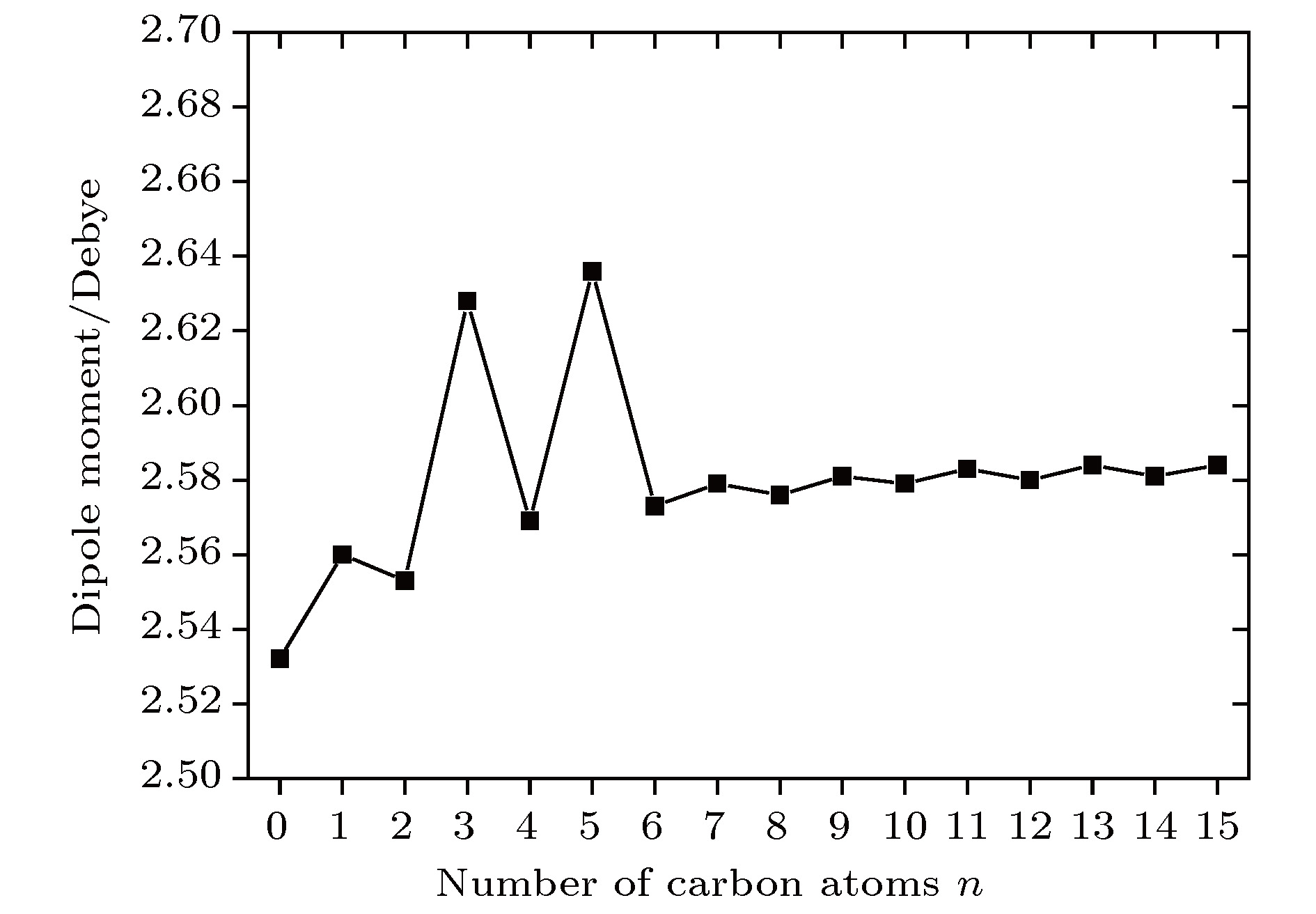
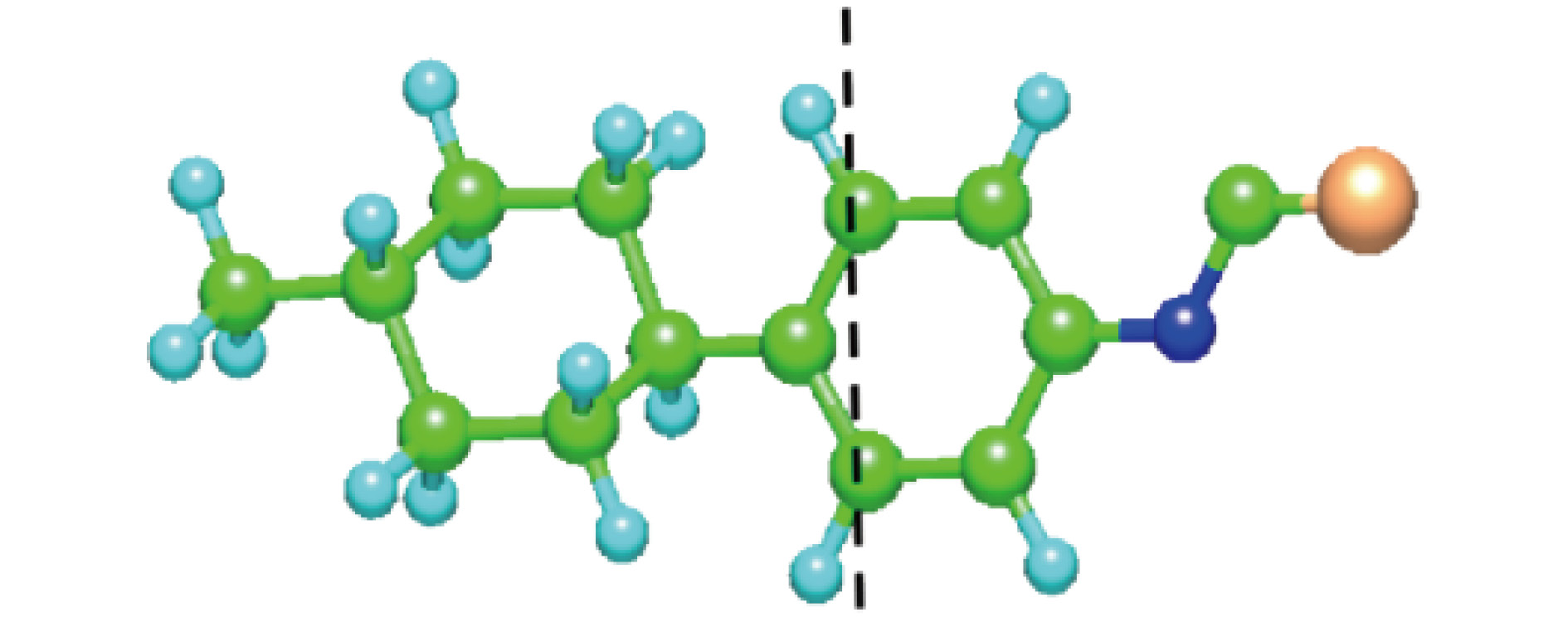
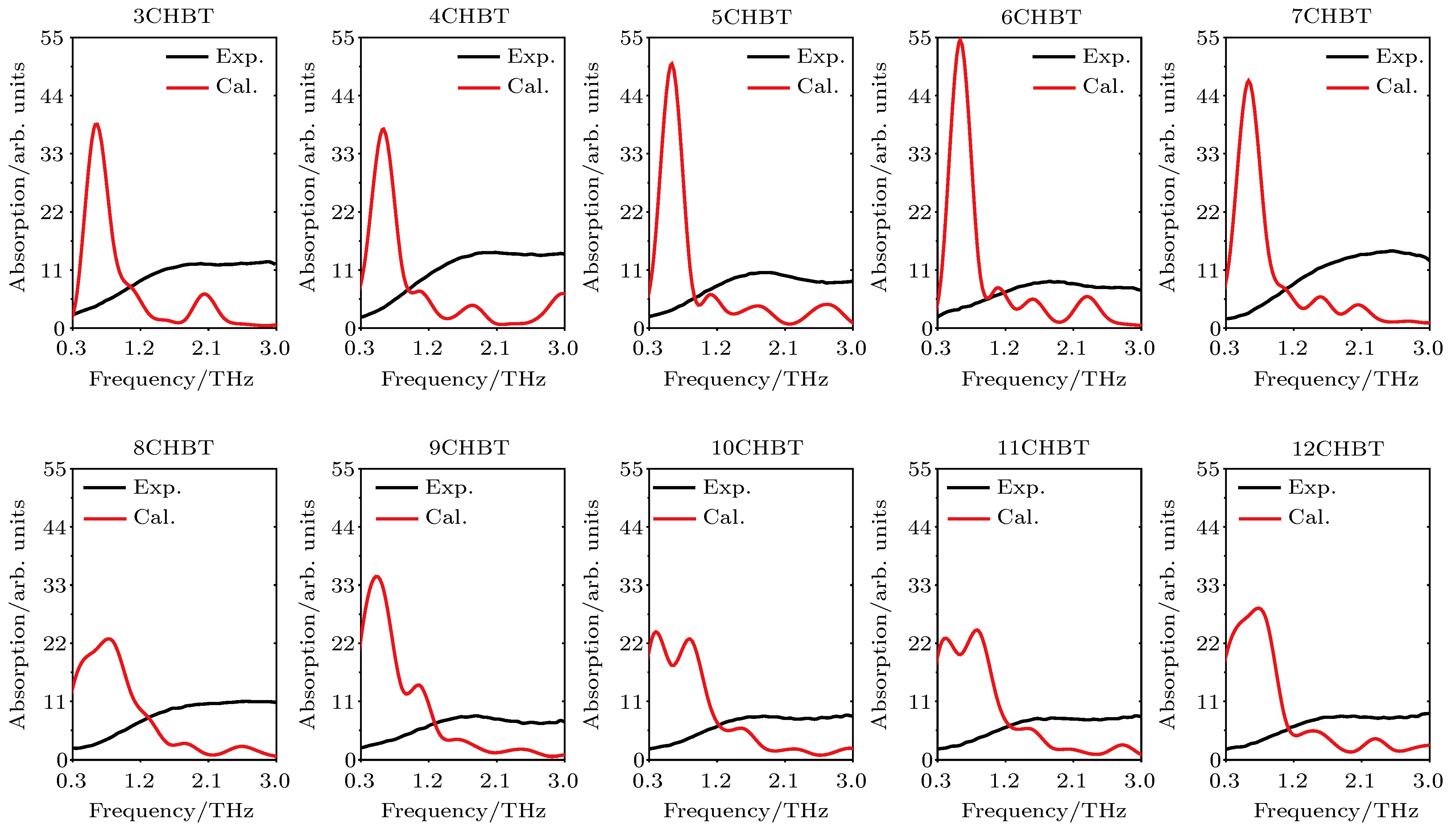
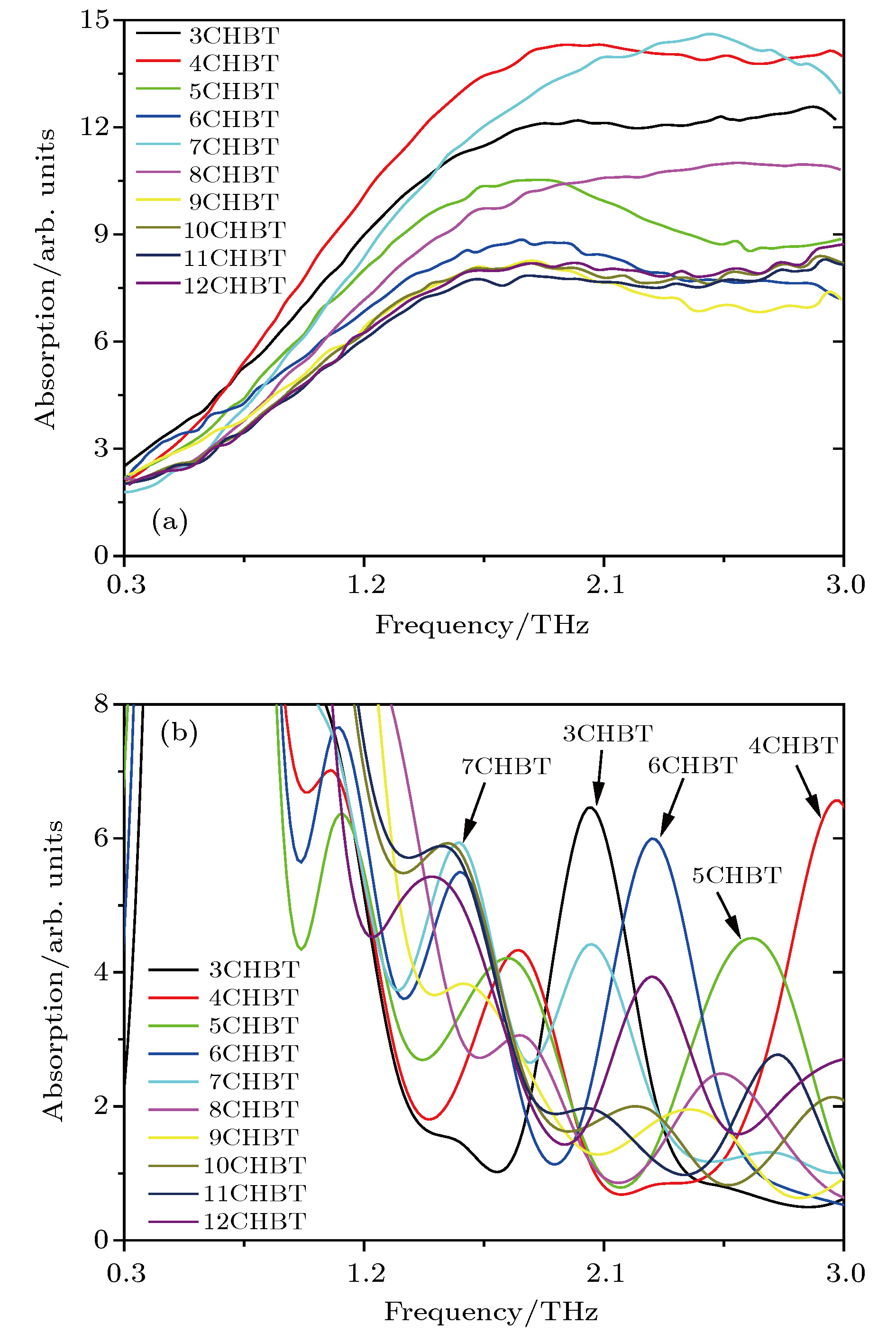
According to density functional theory, in this paper we report a simulation result obtained by using the Gaussian09 package. Adopted in the calculation are an optimized Opt Freq and a base group of B3LYP/6-311g to simulate the absorption of 16 kinds of liquid crystal (LC) molecules of 4-(trans-4-n-alkylcyclohexyl) isothiocyanatobenzenes (CHBT) in a 0.1−5.0 terahertz band (THz). The results show that in the low terahertz band, the absorption is caused mainly by the vibration and rotation of the molecules. So for convenience, we present an novel analytical method of studying the influence of molecular moment of inertia and mass center of gravity shift on absorption. An important result is found that the length of the molecular alkyl chain can lead to different molecular mass, mass center of gravity and moment of inertia, which causes the rotation and vibration of the molecule to be different. These factors lead to the difference in terahertz wave absorption. In the 0.1−5.0 terahertz band, the molecules with 3−7 alkyl chain carbon atoms show a strong absorption. As a reference, reducing and increasing the carbon atoms in the alkyl chain will cause the molecules to reduce the absorption of terahertz waves . In the end, the calculated results are compared with the experimental results obtained from 10 molecules according to the reference data in a frequency range of 0.3−3.0 terahertz. It is found that in the low frequency band there exist some differences between the calculation results and the experimental measurements, in which the difference in the position of the absorption peak may originate from a hydrogen bond. Comparing the relative magnitudes of the absorption intensities, it is found that the experimental measurements are consistent with the calculated results, indicating that the absorption intensity comes from the absorption of dipole vibration and rotation, which demonstrates the positive significance of computational simulation. We look forward to the experimental measurements in the future, and correct the calculation methods and keywords as well as the parameters such as temperature calculation that is to be done in future work. As a theoretical basis, the calculation results can better reflect the absorption of molecular materials, and it is expected to provide useful suggestions for designing and synthesizing the liquid crystal molecules.












 DownLoad:
CSV
DownLoad:
CSV


 DownLoad:
DownLoad:






