The expression of equilibrium pressure tensor for uniform systems derived from Virial theorem can be divided into two parts: dynamic pressure tensor and configuration pressure tensor. The local mean pressure tensor is obtained by physical analysis of the equilibrium non-uniform system. In this paper, this expression is derived in a more concise way. The theoretical relationship, with undetermined parameters, between the three parts (the volume contribution, the surface contribution and the line contribution) of the average configuration pressure of a homogeneous fluid system and the local average size
L
*with the atomic diameter as length unit greater than 8 is given. Taking argon as an example, molecular dynamics simulation is carried out by using the Lennard-Jones potential between atoms at temperature 180 K and density 0.8 (atomic diameter)
–3. The simulation curves of configuration pressure and its three parts are given, and the undetermined coefficients in the theoretical relationship under the condition of
L
*> 8 are determined. It is found that for
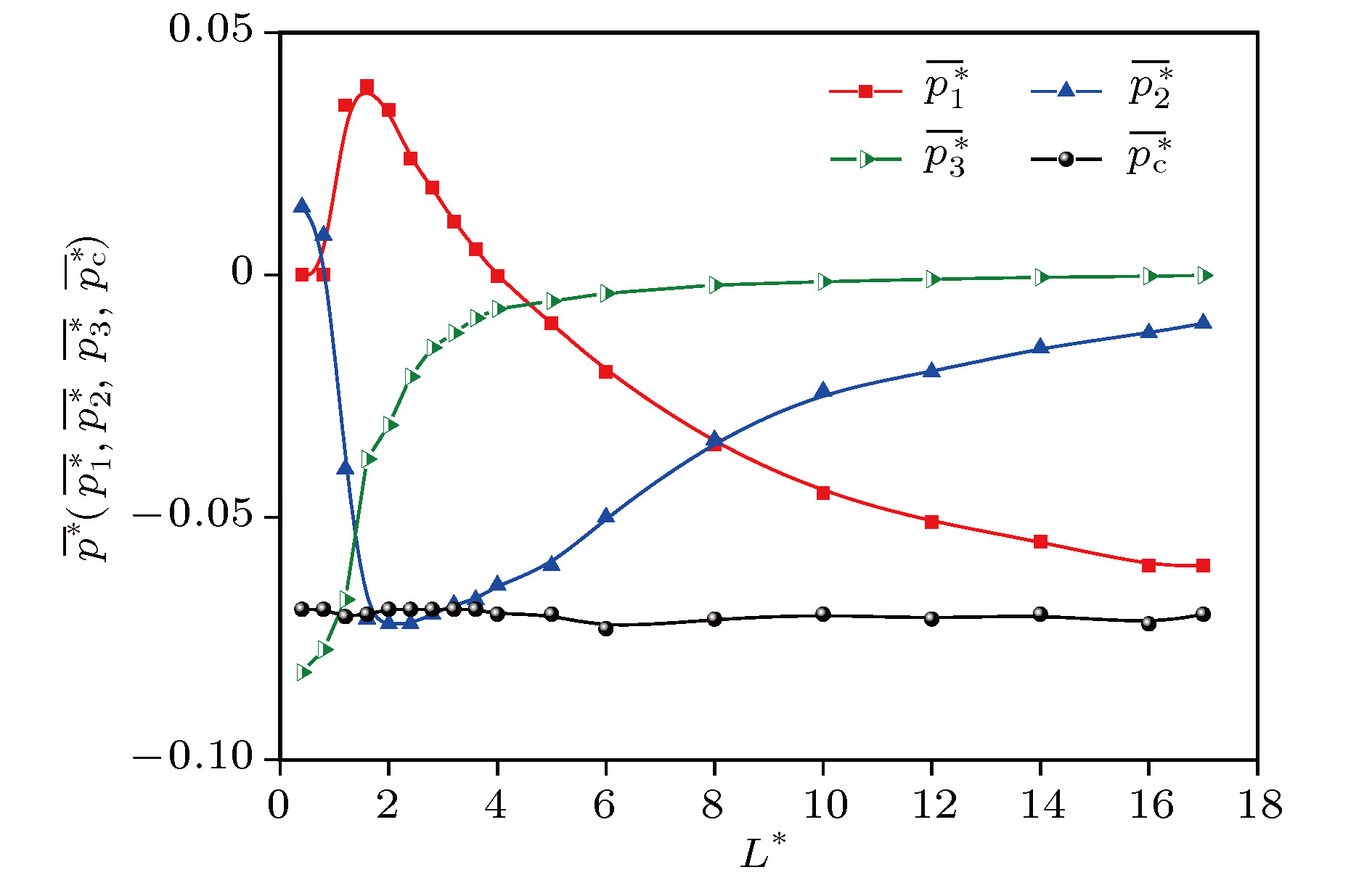
L
*> 2, With the increase of
L
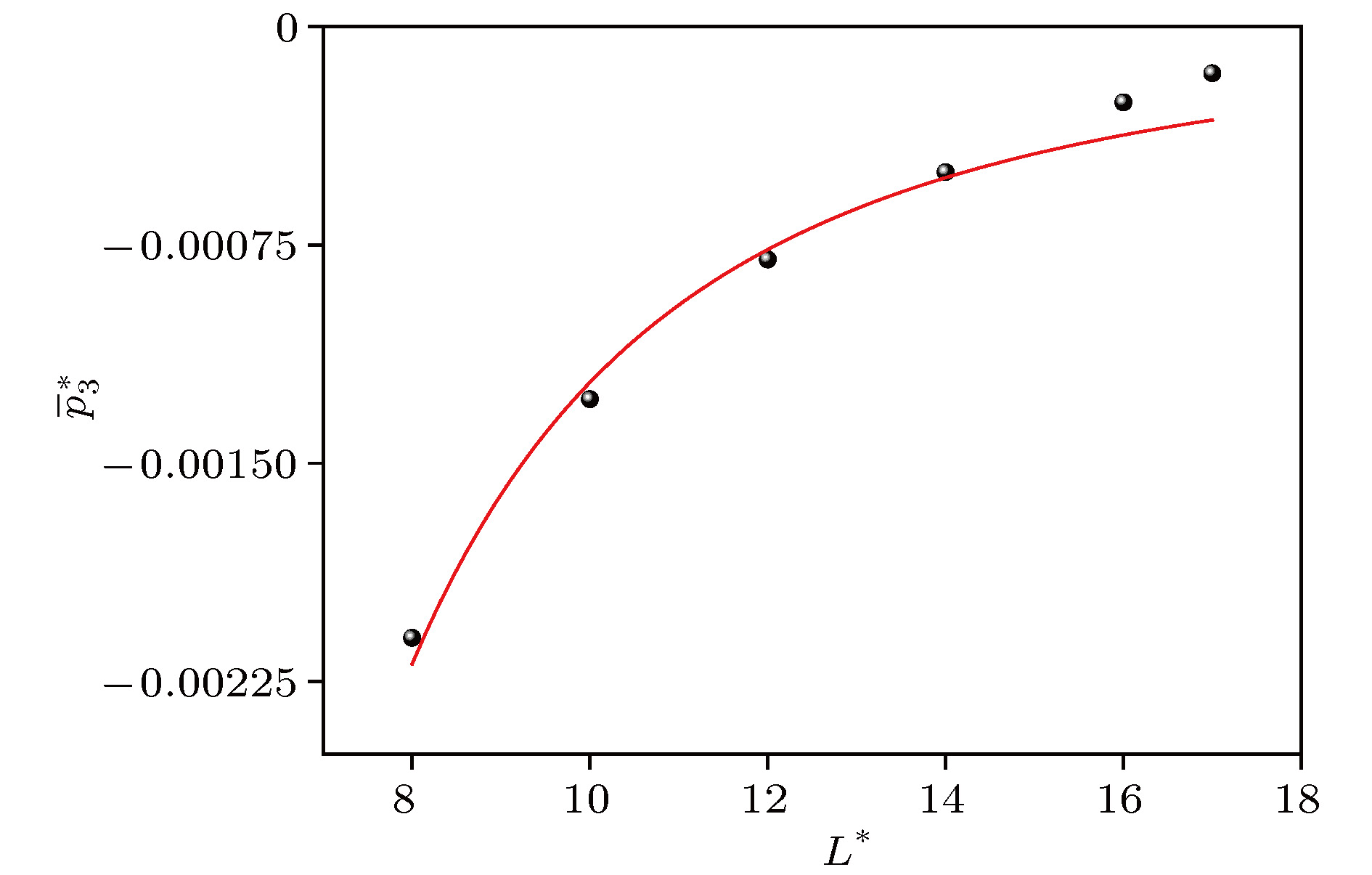
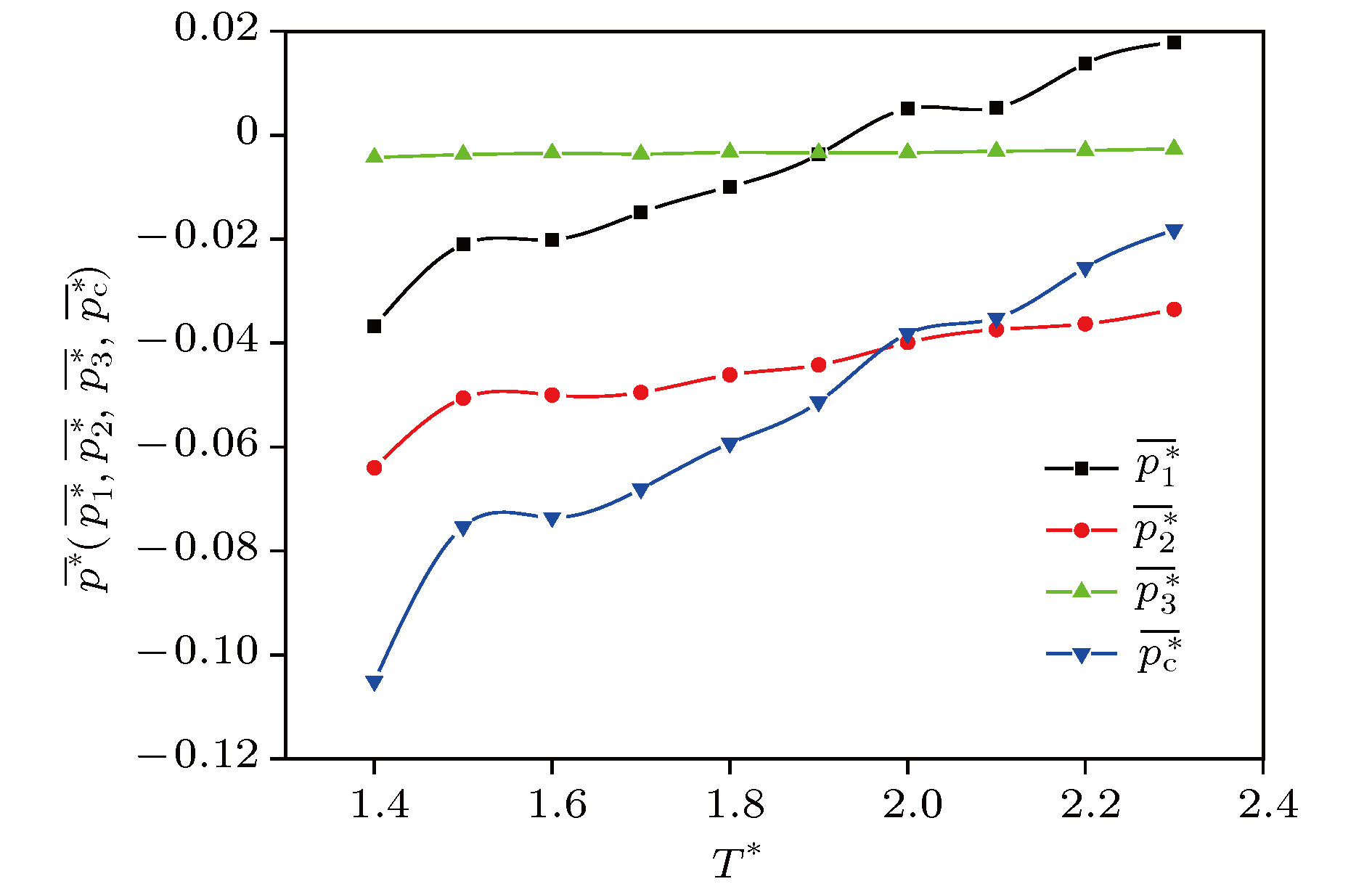
*, the volume contribution decreases monotonously from the positive pressure to the negative total configuration pressure, while the surface contribution and the line contribution both increase monotonously and tend to zero, but the line contribution tends to zero fastest. The complex characteristics of various behaviors are explained physically. It is concluded that the surface contribution and the line contribution can be neglected only if
L
*are large enough. In nanoscale, the neglect of the surface contribution and the line contribution, i.e., the neglect of the boundary effect, will bring obvious errors to the calculation. Finally, molecular dynamics simulation shows that the configuration pressure increases with the increase of temperature. These conclusions are significant for optimizing the selection of pressure tensor in molecular dynamics simulation.













 DownLoad:
CSV
DownLoad:
CSV


 DownLoad:
DownLoad:






