










-
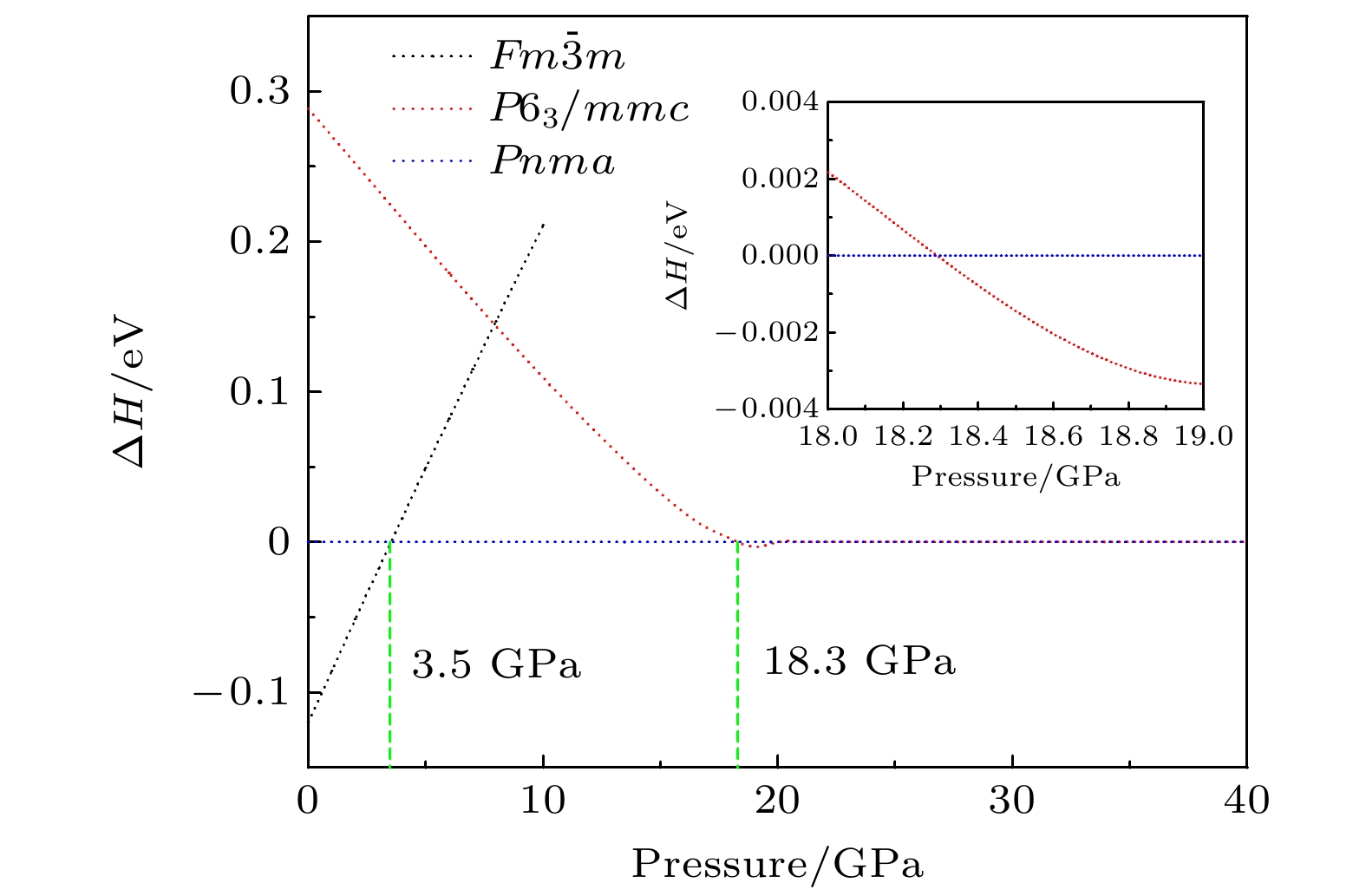
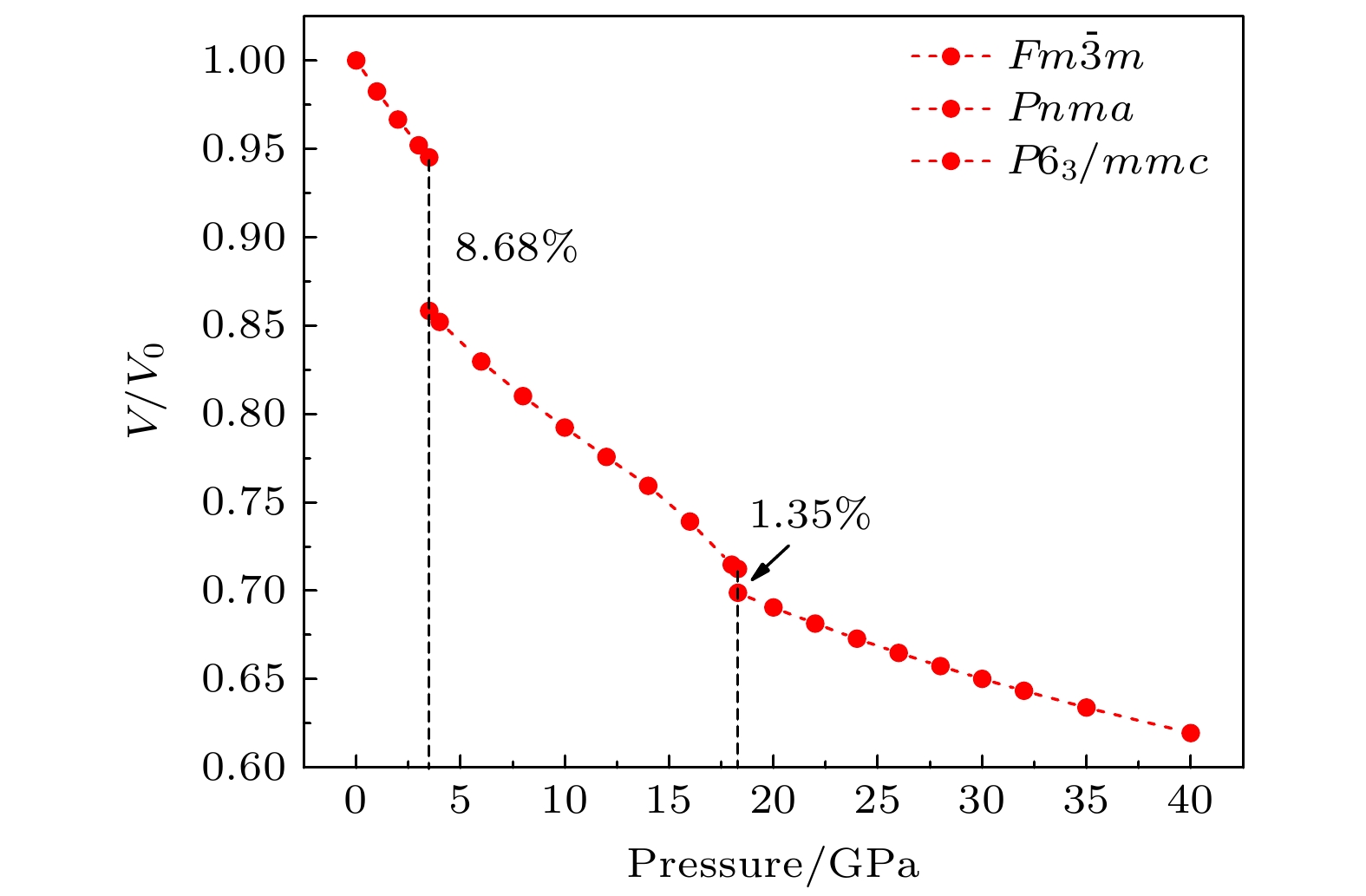
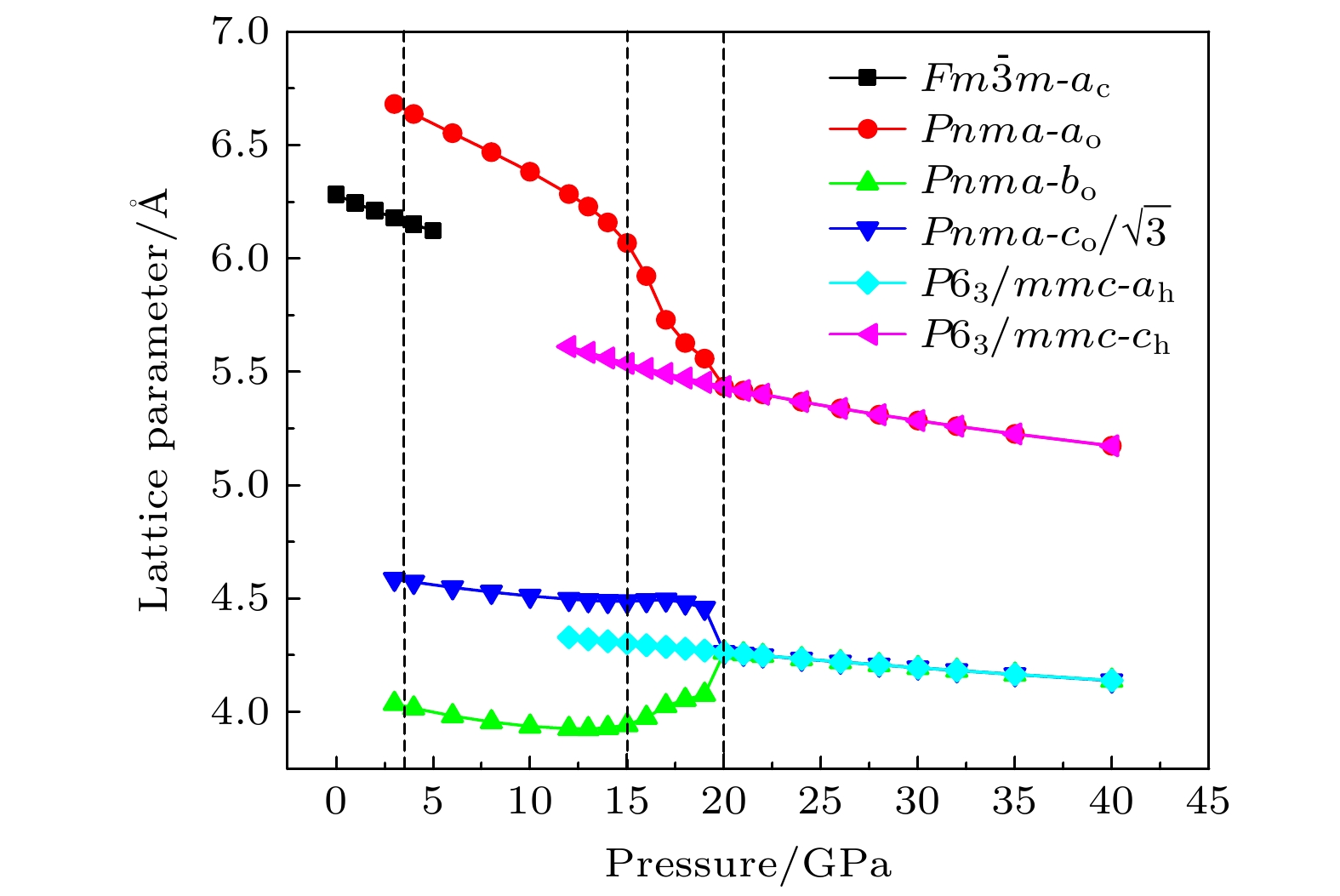
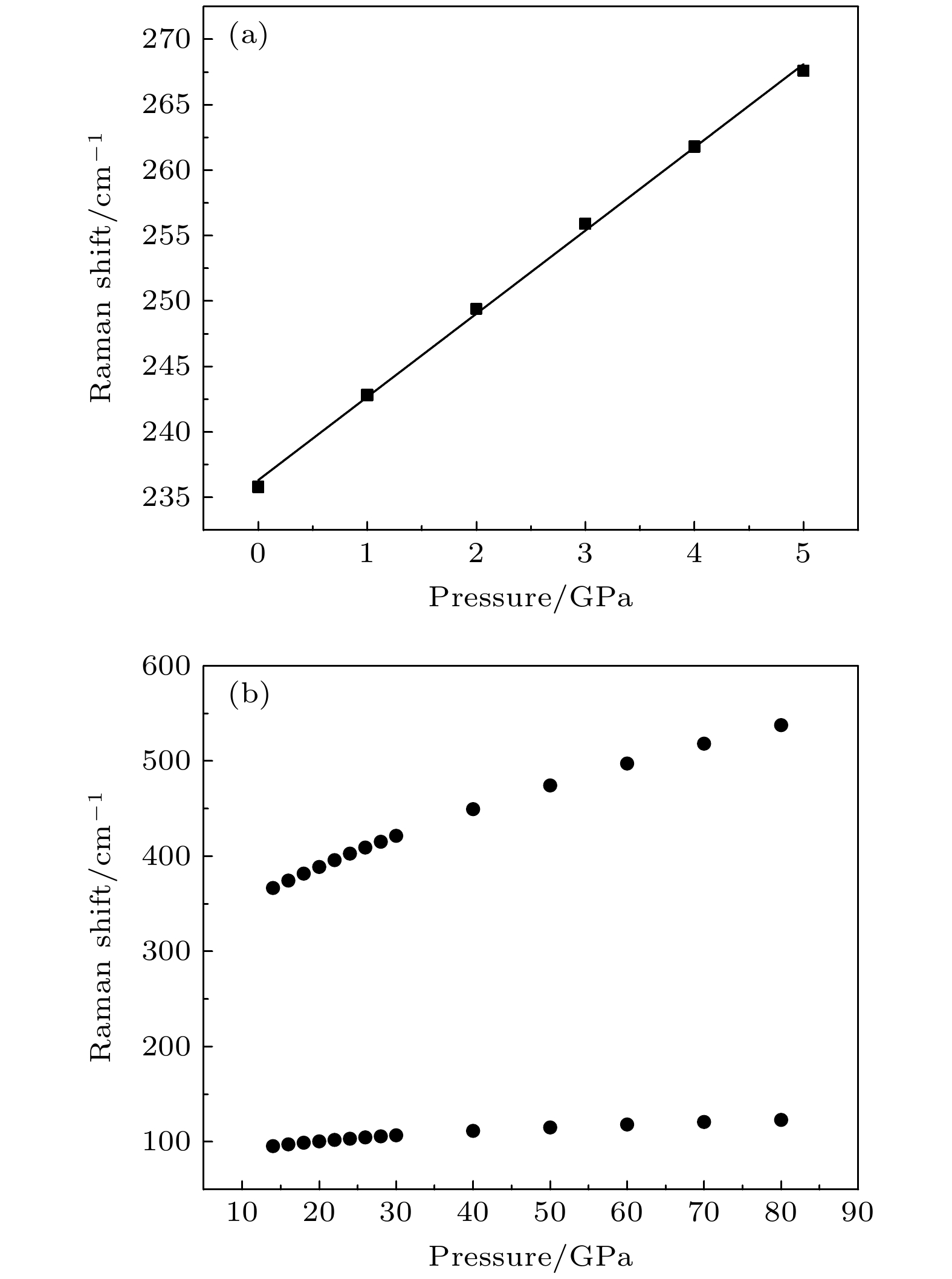
通过理论计算研究了BaF 2在高压下的晶体结构及物理性质. 结果表明, 在3.5和18.3 GPa, BaF 2依次经历了 Fm
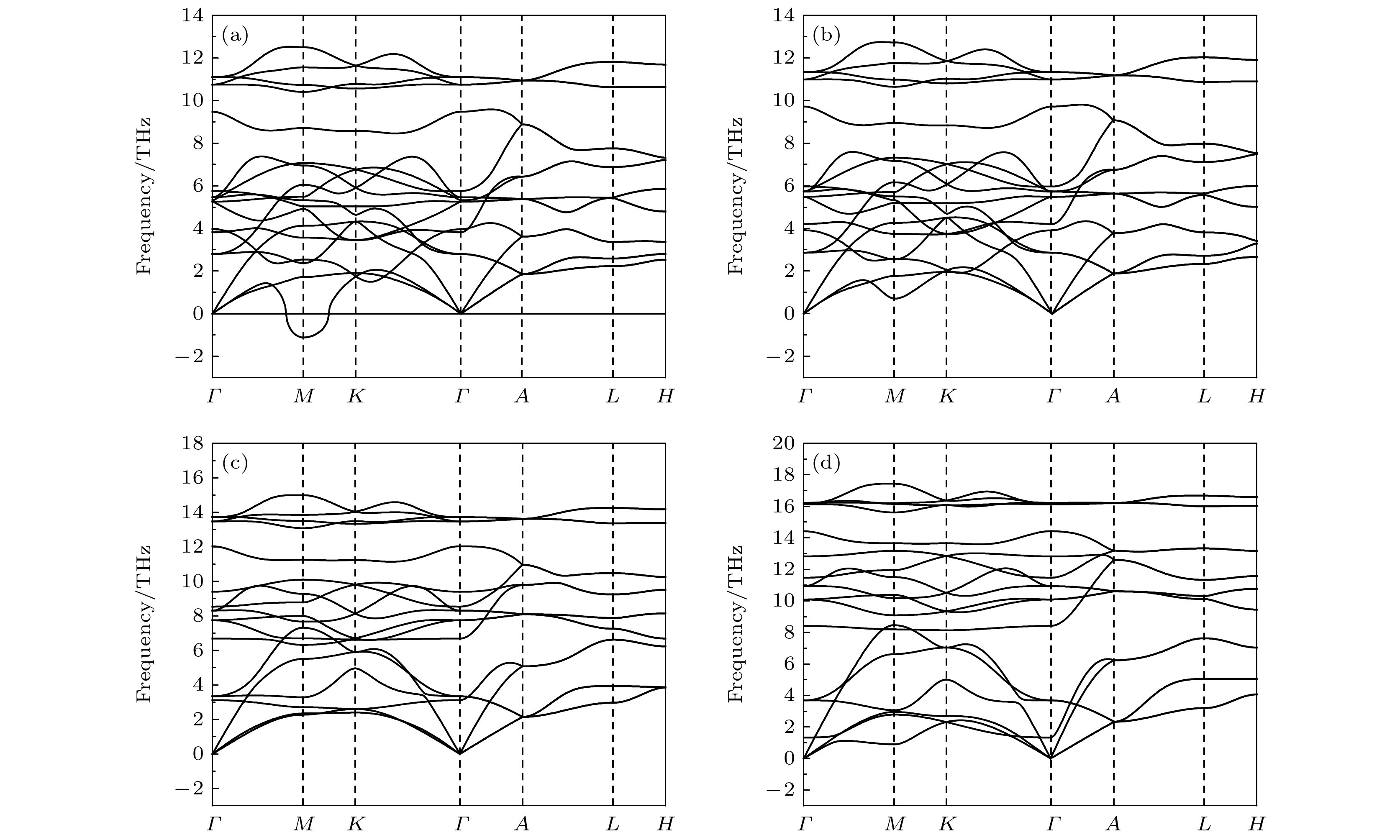
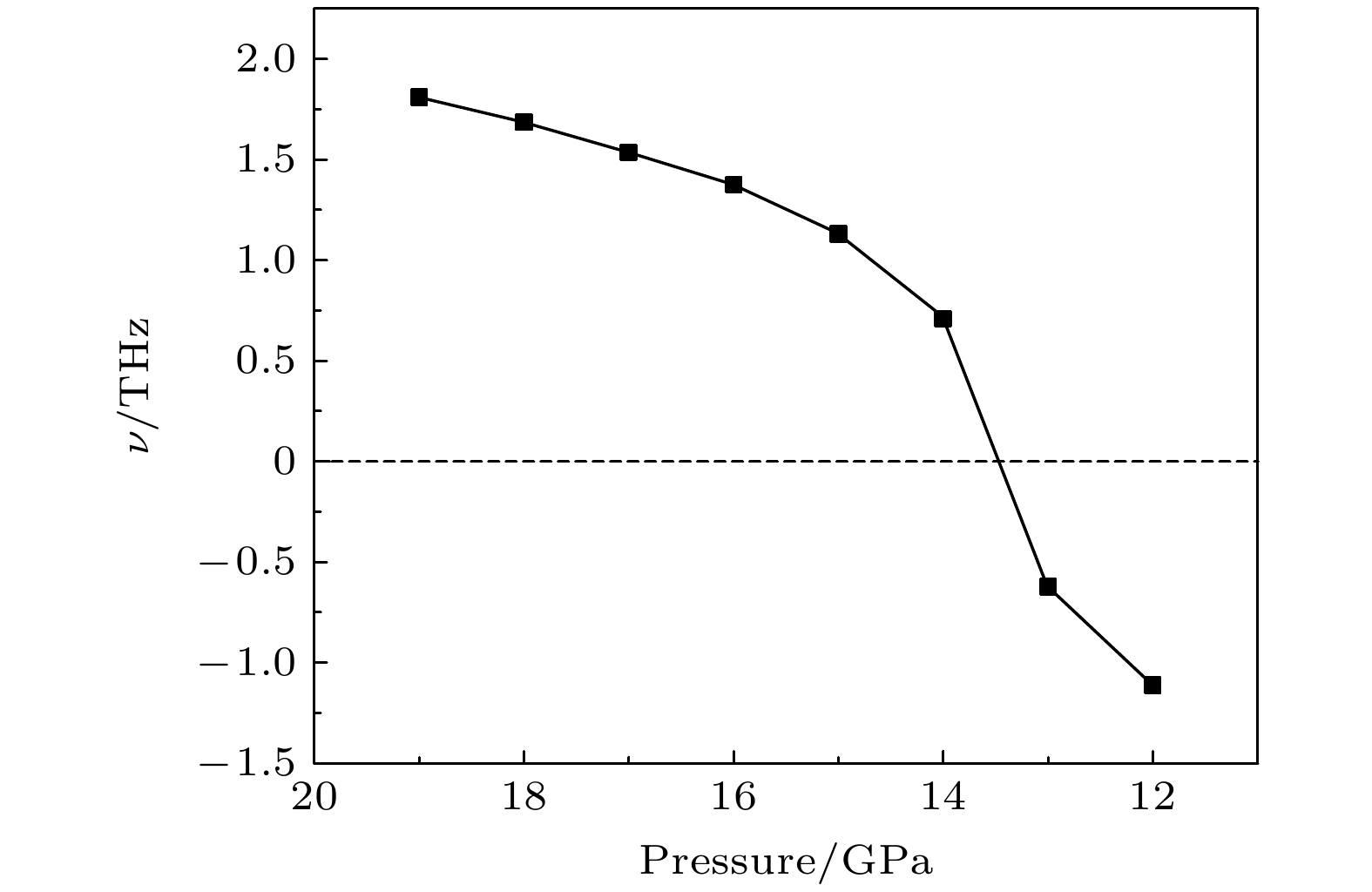
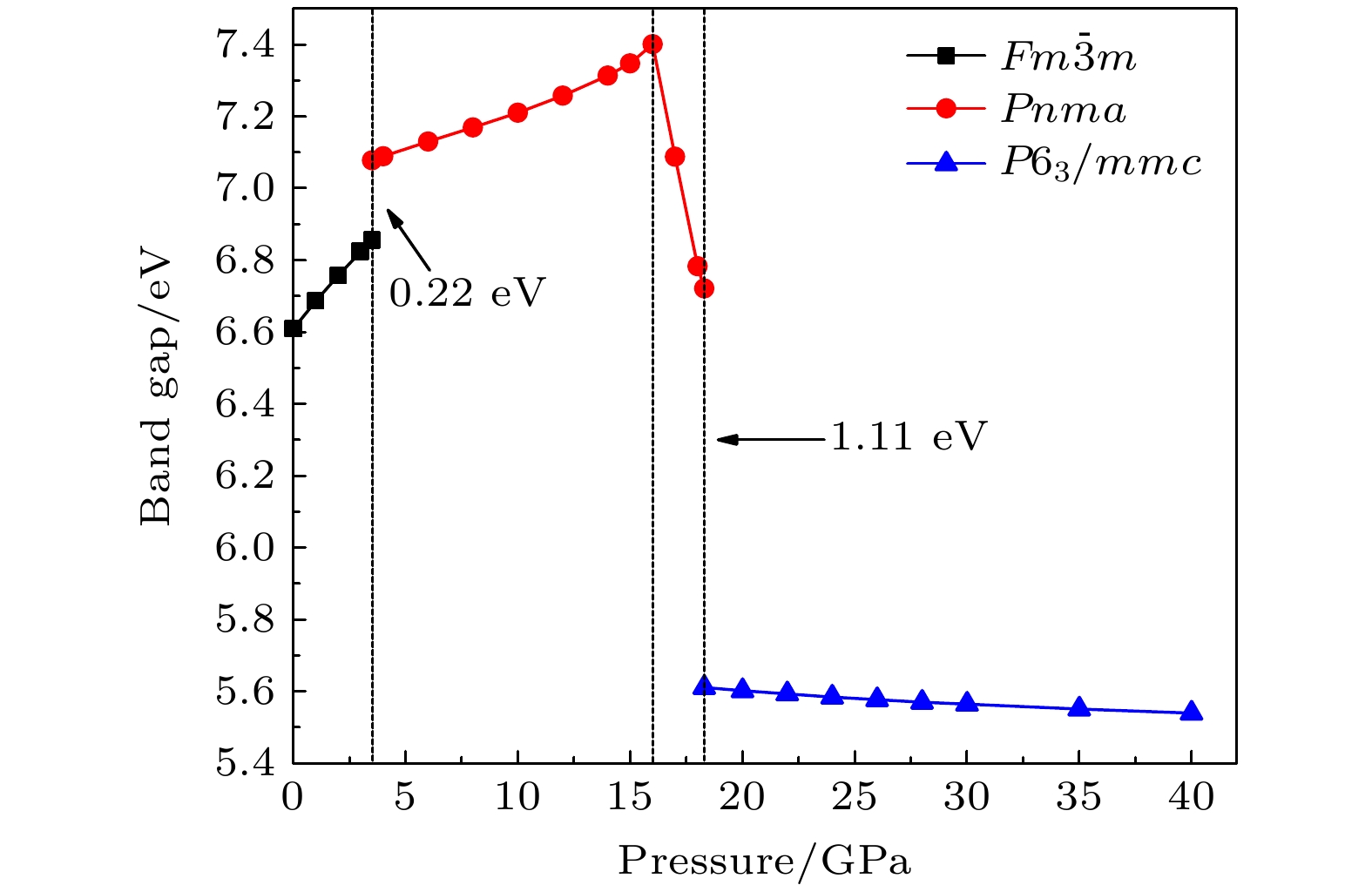
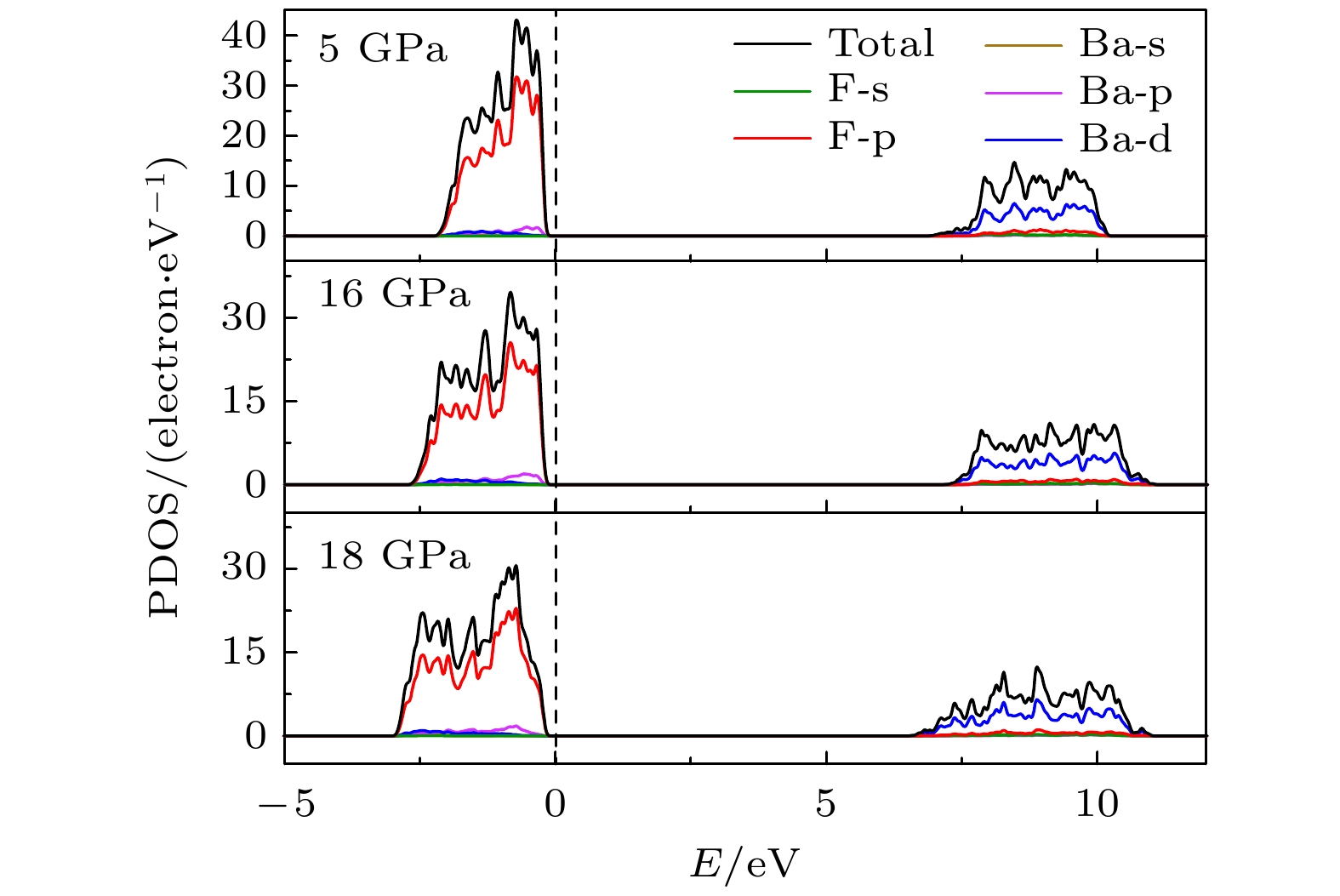
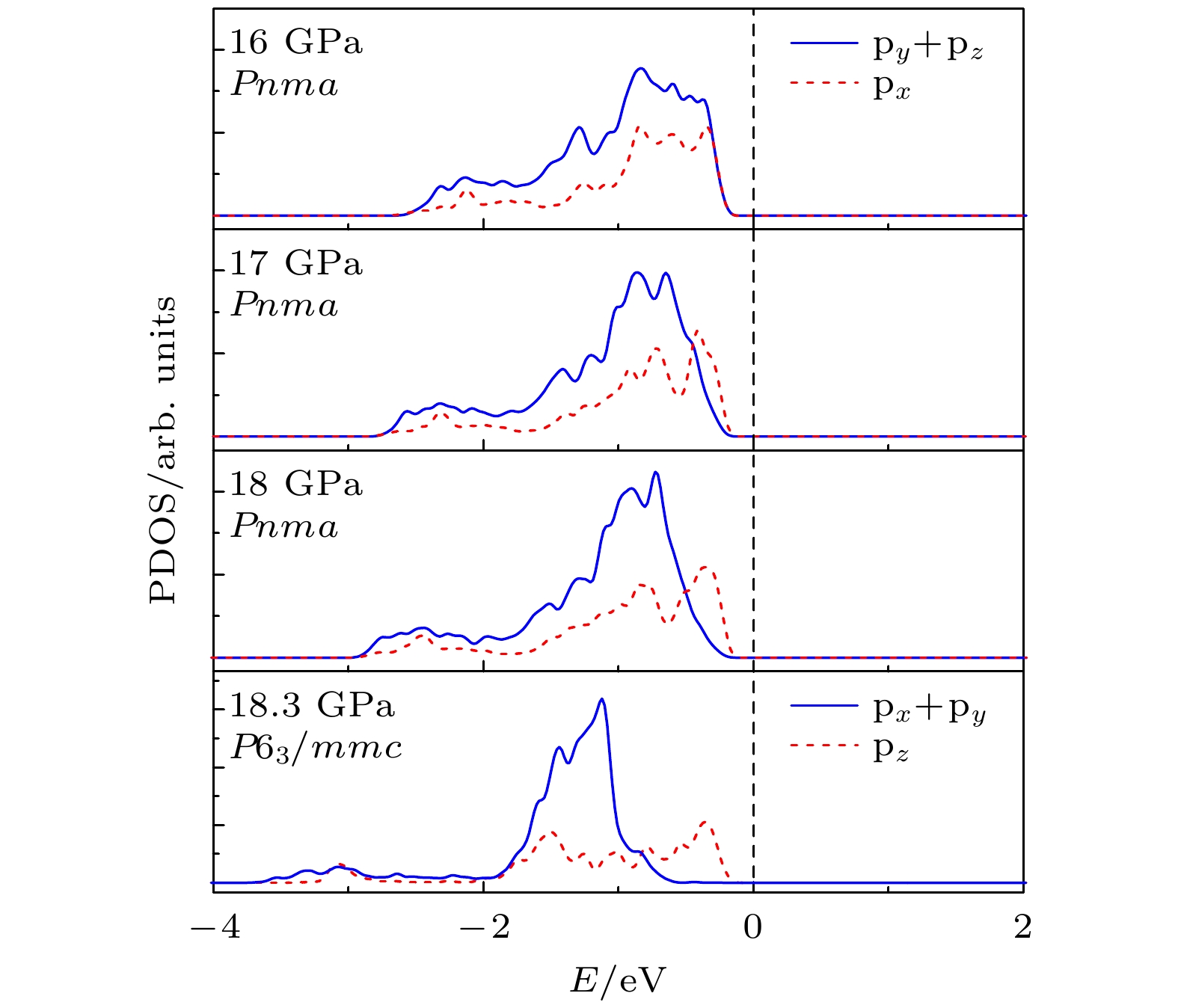
$ \overline {3} $ m- Pnma- P6 3/ mmc两次结构相变, 相变过程伴随着体积的塌缩, 均为一级相变. 约15 GP时, Pnma相晶轴压缩性出现异常, 表现为随压强增大, 晶轴 b o轻微增加, a o略微减小. 对其电子态密度进行分析发现, 在16 GPa以后, 由于F1原子的p y+ p z与p x轨道电子离域, 导致其带隙随压强增加而降低. 在约20 GPa时, Pnma相完全转变为 P6 3/ mmc相, 相变完成. 对BaF 2的拉曼峰位随压强变化进行了计算, 为其高压拉曼光谱行为提供了相应的理论依据. 计算了 P6 3/ mmc相在不同压强下的声子色散曲线, 揭示了其卸压过程中的滞后机制, 计算结果还预测该物相至少可以稳定到80 GPa.There have been some theoretical studies of high pressure phase transition behavior of BaF 2, while in most cases the attention is paid mainly to the optical and electrical properties of BaF 2under increasing pressure. To date, there has been still a lack of theoretical explanation for the hysteresis phenomenon of high-pressure phase of BaF 2when the pressure is released. In addition, the pressure-dependent behavior of the BaF 2band gap is still under controversy, and there are few studies of its high-pressure Raman spectra. Therefore, first principle is used to make a supplementary calculation of the high pressure behavior of BaF 2. For a given pressure Pand temperature T, the thermodynamic stable phase has the lowest Gibbs free energy. The calculations are performed at zero temperature and hence, the Gibbs free energy becomes equal to the enthalpy. Thus, the variation of enthalpy is calculated as a function of pressure to study the high-pressure phase stability of BaF 2based on density functional theory as implemented in the Vienna ab initio simulation package (VASP). The results show that the BaF 2undergoes two structural phase transitions from Fm3 m(cubic) to Pnma(orthorhombic) and then to P6 3/ mmc(hexagonal) with increasing pressure, and their corresponding transition pressures are 3.5 and 18.3 GPa, respectively. By calculating the evolution of lattice constant with pressure, it is found that at about 15 GPa (near the second phase transition pressure), the lattice constants of the Pnmastructure show abnormal behavior (a slight increase in b oand a slight decrease in a o). We suggest that this behavior leads the band gap to decrease, indicated by analyzing the calculated results of Pnmastructure of other materials. The Pnmastructure completely transforms into P6 3/ mmcstructure at about 20 GPa. By analyzing the phonon dispersion curves of BaF 2as a function of pressure, the structural stability information of the material can also be obtained. Then the density functional perturbation theory (DFPT) is used to calculate the phonon dispersion curves of BaF 2by VASP code and Phonopy code. The hysteresis phenomenon of the P6 3/ mmcstructure, when the pressure is released, is explained by the kinetic stability. The results predict that the P6 3/ mmcstructure can be stabilized at least to 80 GPa.-
Keywords:
- first-principles/
- BaF2/
- phase transition/
- hysteresis
[1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] [21] [22] [23] [24] [25] [26] [27] [28] [29] [30] [31] [32] [33] [34] [35] [36] [37] [38] [39] [40] [41] [42] [43] [44] [45] -
Mode ω/cm–1 Mode ω/cm–1 Mode ω/cm–1 Ag 81.2 Ag 190.6 B2g 268.7 B3g 81.4 B1g 203.3 Ag 283.3 B1g 90.2 Ag 211.2 B1g 304.4 Ag 112.9 B3g 218.7 B3g 309.6 B2g 151.5 B2g 224.0 Ag 321.6 B2g 174.7 B2g 251.2 B2g 363.8  下载:
导出CSV
下载:
导出CSV
-
[1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] [21] [22] [23] [24] [25] [26] [27] [28] [29] [30] [31] [32] [33] [34] [35] [36] [37] [38] [39] [40] [41] [42] [43] [44] [45]


 下载:
下载:
计量
- 文章访问数:4377
- PDF下载量:123
- 被引次数:0