










-
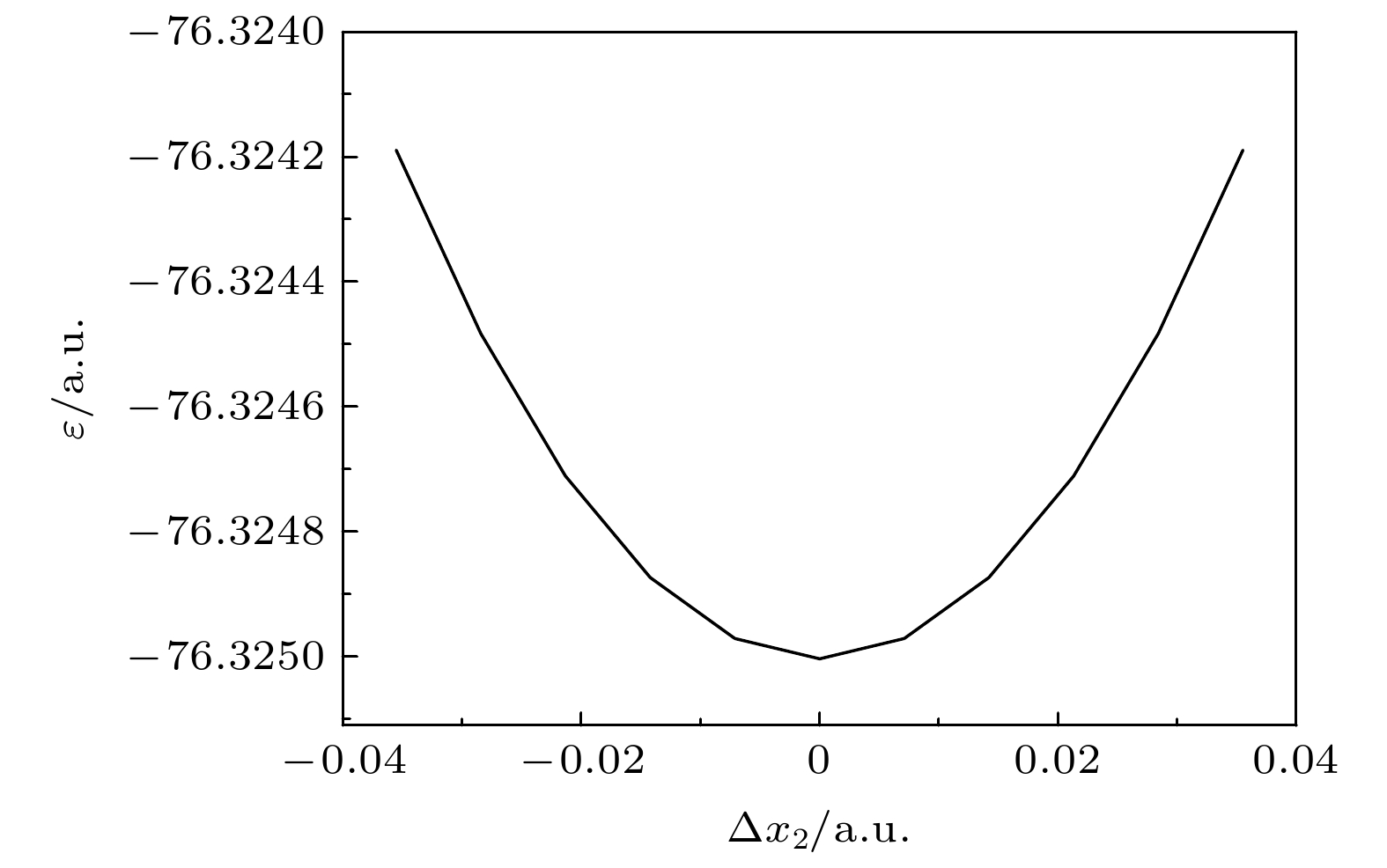
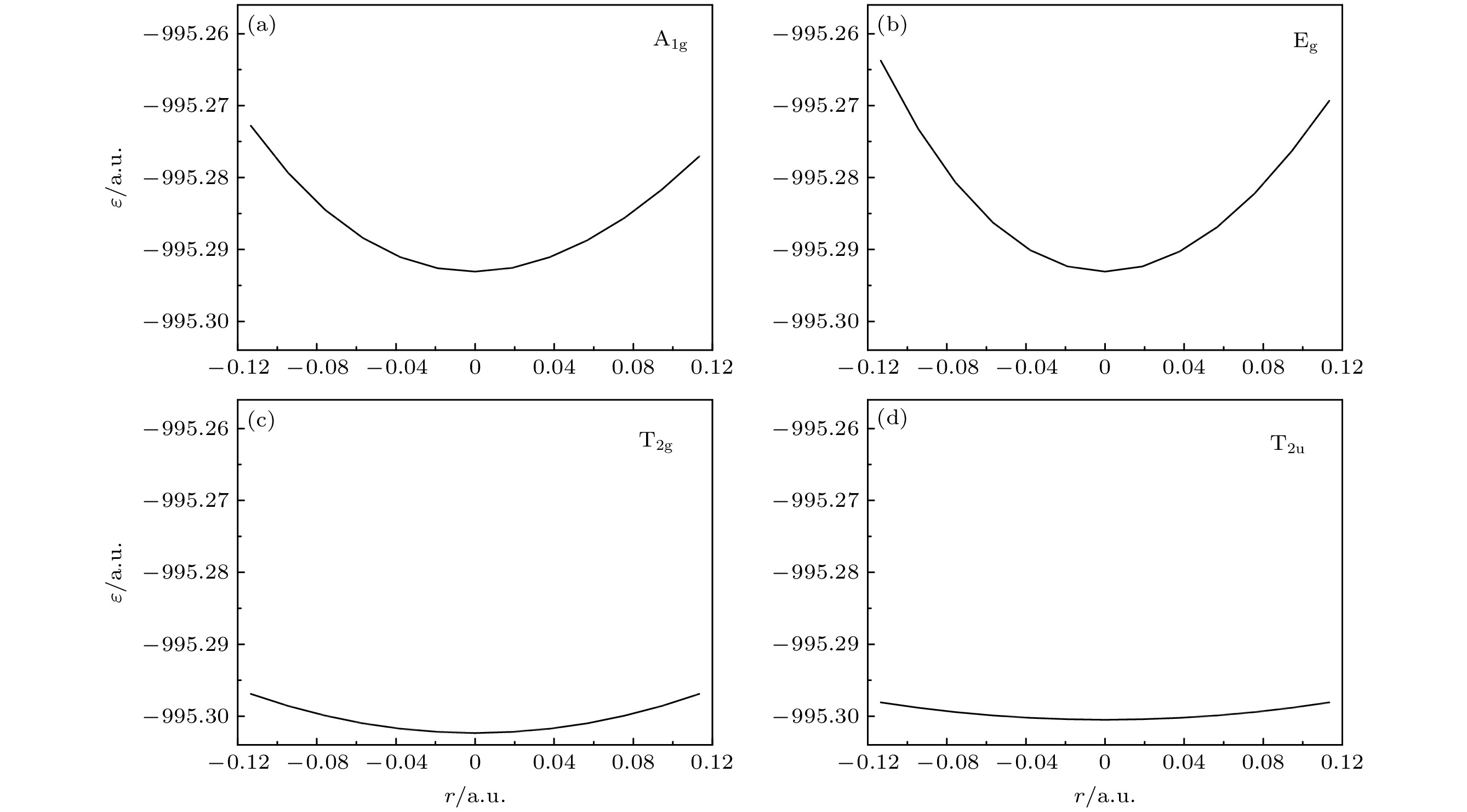
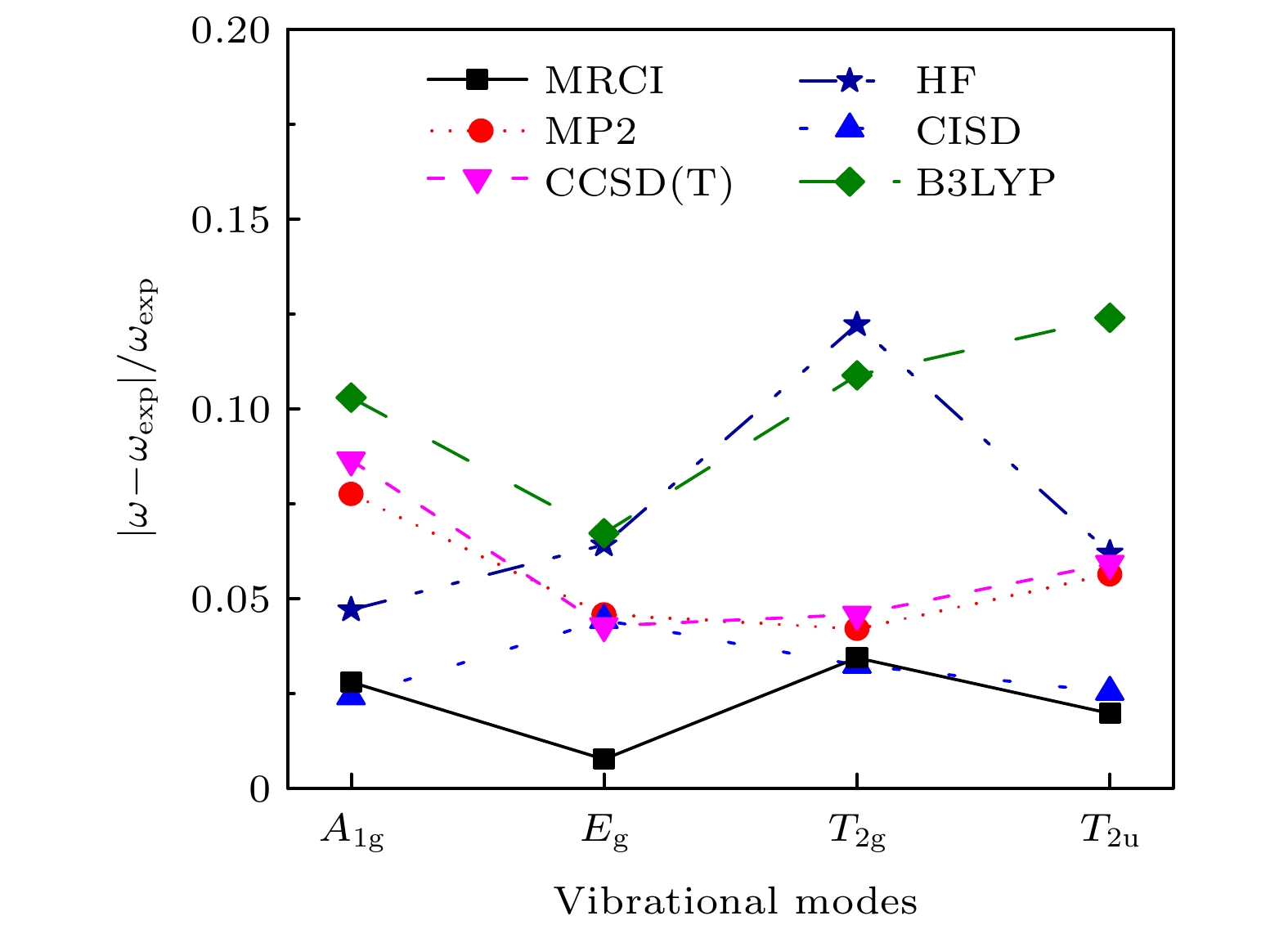
针对较大分子振动频率的量化计算, 提出了一个节省计算成本的方法. 含 N个原子的分子的振动频率的计算通常需要计算3 N维势能超曲面及其二阶导数构成的Hessian矩阵, 然后解其特征方程得到全部简正振动模式的振动频率. N越大, 计算成本越大. 本文提出, 针对那些由平衡结构和对称性就能完全确定的振动模式, 可以逐个计算其振动频率. 当仅考虑一个振动模式时, 3 N维的Hessian矩阵的计算转化为一维的势能曲线的计算. 基于简谐振子近似推导单一振动模式下分子势能曲线的表达式, 接着量化计算势能曲线, 将势能曲线拟合到表达式中以获得振动频率. 相比计算3 N维势能超曲面及其二阶导数的Hessian矩阵, 仅计算一维势能曲线而节省下来的计算资源可以允许选择更高级别的计算方法和采用更为完备的基组, 提高计算的精度. 本文首先以计算水分子的 B 2振动模式的振动频率为例, 说明了这种方法的可行性. 接着将这种方法应用到SF 6分子中. 多参考组态相互作用(MRCI)方法是计算电子相关能的有效方法, 本文采用MRCI/6-311G*基组分别计算了SF 6的A 1g, E g, T 2g和T 2u四个振动模式的振动频率, 通过与其他方法的结果以及实验结果相比较, 本文计算的四个频率的相对误差最小.Quantum calculation of molecular vibrational frequency is important in investigating infrared spectrum and Raman spectrum. In this work, a low computational cost method of calculating the quantum chemistry of vibrational frequencies for large molecules is proposed. Usually, the calculation of vibrational frequency of a molecule containing Natoms needs to deal with the Hessian matrix, which consists of second derivatives of the 3 N-dimensional potential hypersurface, and then solve secular equations of the matrix to obtain normal vibration modes and the corresponding frequencies. Larger Nimplies higher computational cost. Therefore, for a limited computational hardware condition, higher-level computations for large Natomic molecule’s vibrational frequencies cannot be implemented in practice. Here we solve this problem by calculating the vibrational frequency for only one vibrational mode each time instead of calculating the Hessian matrix to obtain all vibrational frequencies. When only one vibrational mode is taken into consideration, the molecular potential hypersurface can be transformed into one-dimensional curve. Hence, we can calculate the curve with high-level computational method, then deduce the expression of one-dimensional curve by using harmonic oscillating approximation and obtain the vibrational frequency by using the expression to fit the curve. It should be noted that this method is applied to vibrational modes whose vibrational coordinates can be completely determined by equilibrium geometry and the molecular symmetry and be independent of the molecular force constants. It requires that there exists no other vibrational mode with the same symmetry but with different frequencies. The lower computational cost for a one-dimensional potential curve than that for 3 N-dimensional potential hypersurface’s second derivatives permits us to use higher-level method and larger basis set for a given computational hardware condition to achieve more accurate results. In this paper we take the calculation of B 2vibrational frequency of water molecule for example to illustrate the feasibility of this method. Furthermore, we use this method to deal with the SF 6molecule. It has 7 atoms and 70 electrons, hence there exists a large amount of electronic correlation energy to be calculated. The MRCI is an effective method to calculate the correlation energy. But by now no MRCI result of SF 6vibrational frequencies has been reported. So here we use MRCI/6-311G* to calculate the potential curves of A 1g, E g, T 2gand T 2uvibrational modes separately, deduce their expressions, then use the expressions to fit the curves, and finally obtain the vibrational frequencies. The results are then compared with those obtained by other theoretical methods including HF, MP2, CISD, CCSD(T) and B3LYP methods through using the same 6-311G* basis set. It is shown that the relative error to experimental result of the MRCI method is the least in the results from all these methods.
[1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] [21] [22] -
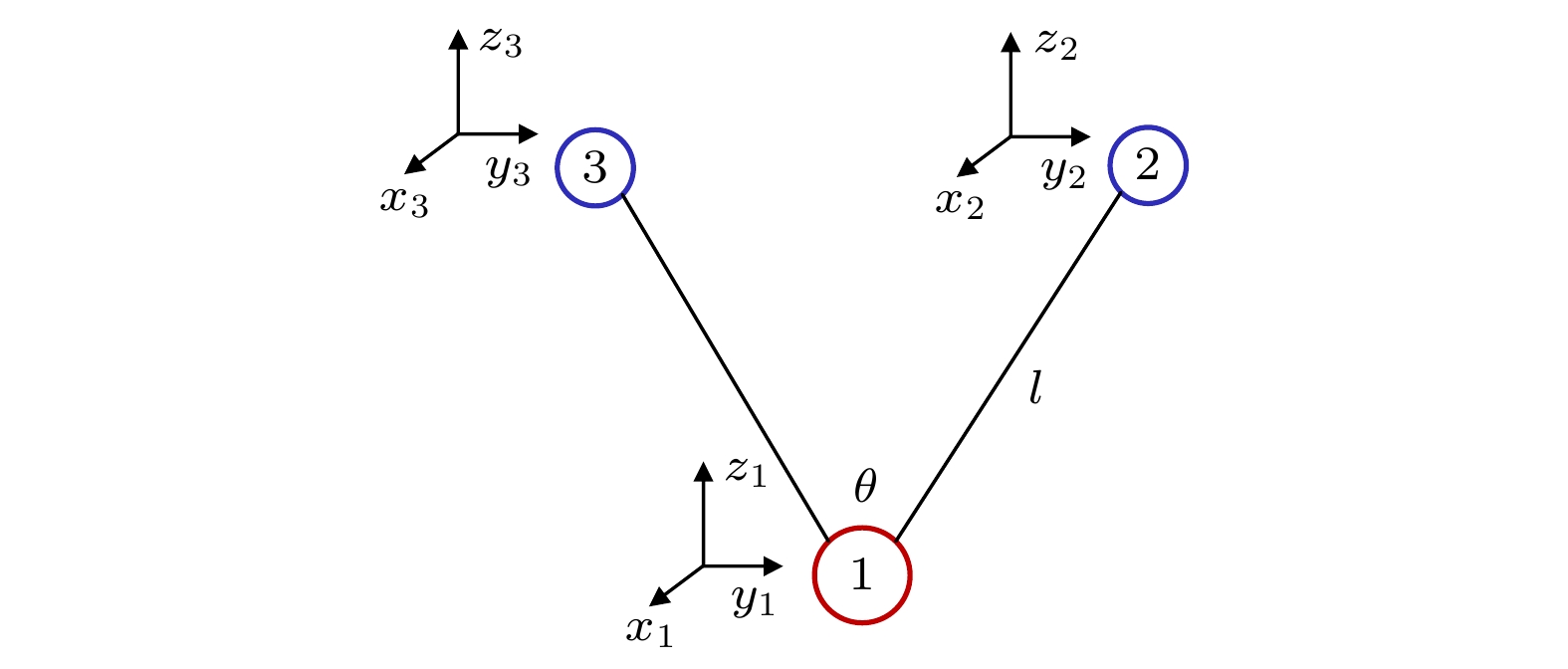
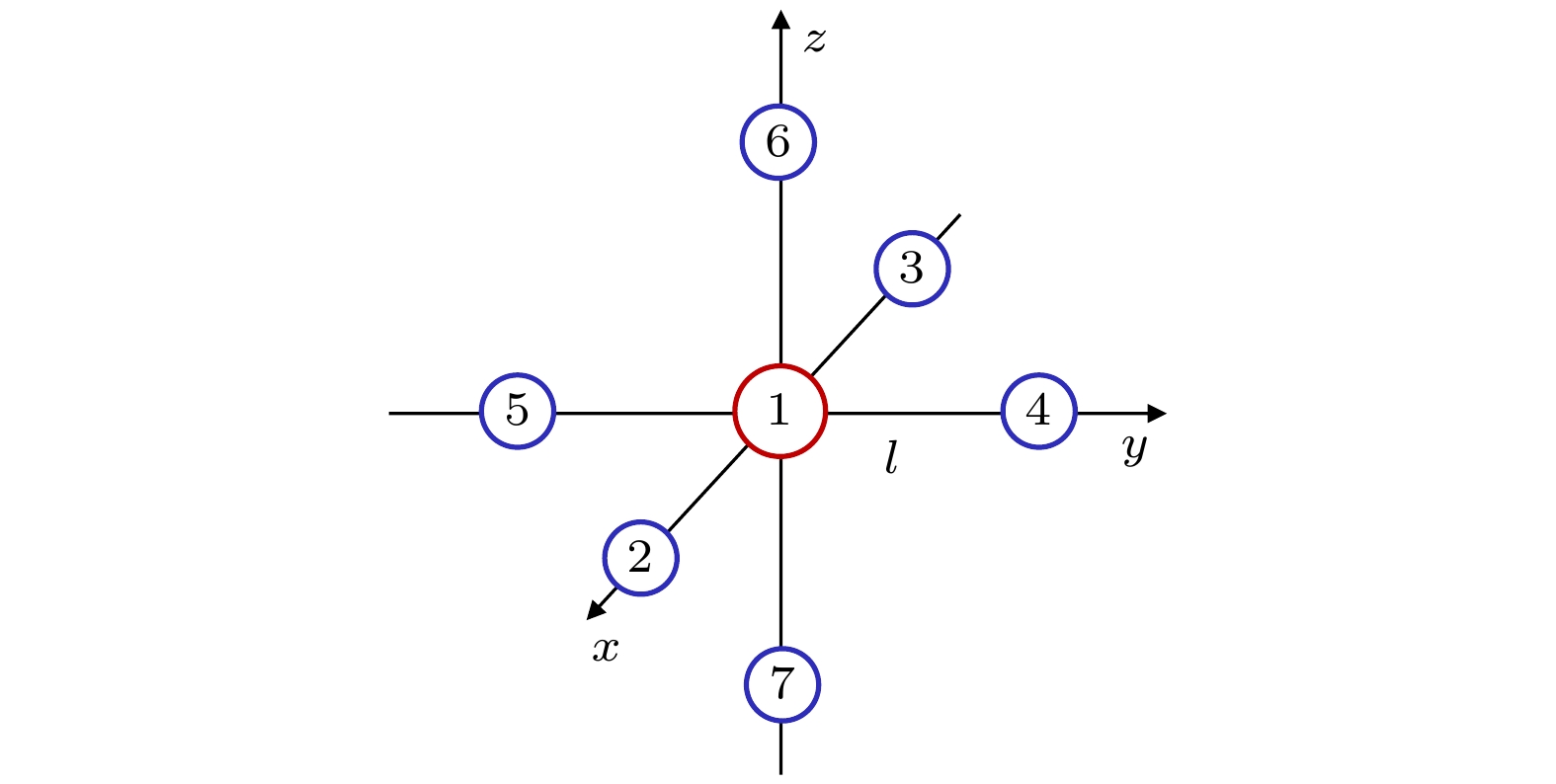
位移坐标 $ {\mathrm{A}}_{1\mathrm{g}} $ $ {\mathrm{E}}_{\mathrm{g}} $ $ {\mathrm{E}}_{\mathrm{g}} $ $ {\mathrm{T}}_{2\mathrm{g}} $ $ {\mathrm{T}}_{2\mathrm{g}} $ $ {\mathrm{T}}_{2\mathrm{g}} $ $ {\mathrm{T}}_{2\mathrm{u}} $ $ {\mathrm{T}}_{2\mathrm{u}} $ $ {\mathrm{T}}_{2\mathrm{u}} $ $ {\Delta x}_{1}, \Delta {y}_{1}, \Delta {z}_{1} $ $ \mathrm{0, 0}, 0 $ $ \mathrm{0, 0}, 0 $ $ \mathrm{0, 0}, 0 $ $ \mathrm{0, 0}, 0 $ $ \mathrm{0, 0}, 0 $ $ \mathrm{0, 0}, 0 $ $ \mathrm{0, 0}, 0 $ $ \mathrm{0, 0}, 0 $ $ \mathrm{0, 0}, 0 $ $ {\Delta x}_{2}, \Delta {y}_{2}, \Delta {z}_{2} $ $ r, \mathrm{0, 0} $ ${r}, \mathrm{0, 0}$ $ -r, \mathrm{0, 0} $ $ 0, r, 0 $ $ \mathrm{0, 0}, r $ $ \mathrm{0, 0}, 0 $ $ 0, r, 0 $ $ \mathrm{0, 0}, 0 $ $ \mathrm{0, 0}, r $ $ {\Delta x}_{3}, {\Delta y}_{3}, \Delta {z}_{3} $ $ -r, \mathrm{0, 0} $ $ -r, \mathrm{0, 0} $ $ \mathrm{r}, \mathrm{0, 0} $ $ 0, -r, 0 $ $ \mathrm{0, 0}, -r $ $ \mathrm{0, 0}, 0 $ $ 0, r, 0 $ $ \mathrm{0, 0}, 0 $ $ \mathrm{0, 0}, r $ $ {\Delta x}_{4}, \Delta {y}_{4}, \Delta {z}_{4} $ $ 0, r, 0 $ $ \mathrm{0, 0}, 0 $ $ \mathrm{0, 2}r, 0 $ $ r, \mathrm{0, 0} $ $ \mathrm{0, 0}, 0 $ $ \mathrm{0, 0}, r $ $ \mathrm{0, 0}, 0 $ $ r, \mathrm{0, 0} $ $ \mathrm{0, 0}, -r $ $ \Delta {x}_{5}, \Delta {y}_{5}, \Delta {z}_{5} $ $0,-r, 0$ $ \mathrm{0, 0}, 0 $ $ 0, -2 r, 0 $ $ -r, \mathrm{0, 0} $ $ \mathrm{0, 0}, 0 $ $ \mathrm{0, 0}, -r $ $ \mathrm{0, 0}, 0 $ $ r, \mathrm{0, 0} $ $ \mathrm{0, 0}, -r $ $ {\Delta x}_{6}, \Delta {y}_{6}, \Delta {z}_{6} $ $ \mathrm{0, 0}, r $ $ \mathrm{0, 0}, -r $ $ \mathrm{0, 0}, -r $ $ \mathrm{0, 0}, 0 $ $ r, \mathrm{0, 0} $ $ 0, r, 0 $ $ 0, -r, 0 $ $ -r, \mathrm{0, 0} $ $ \mathrm{0, 0}, 0 $ $ {\Delta x}_{7}, \Delta {y}_{7}, \Delta {z}_{7} $ $ \mathrm{0, 0}, -r $ $ \mathrm{0, 0}, r $ $ \mathrm{0, 0}, r $ $ \mathrm{0, 0}, 0 $ $ -r, \mathrm{0, 0} $ $ 0, -r, 0 $ $ 0, -r, 0 $ $ -r, \mathrm{0, 0} $ $ \mathrm{0, 0}, 0 $  下载:
导出CSV
下载:
导出CSV
MRCI/6-311G* HF
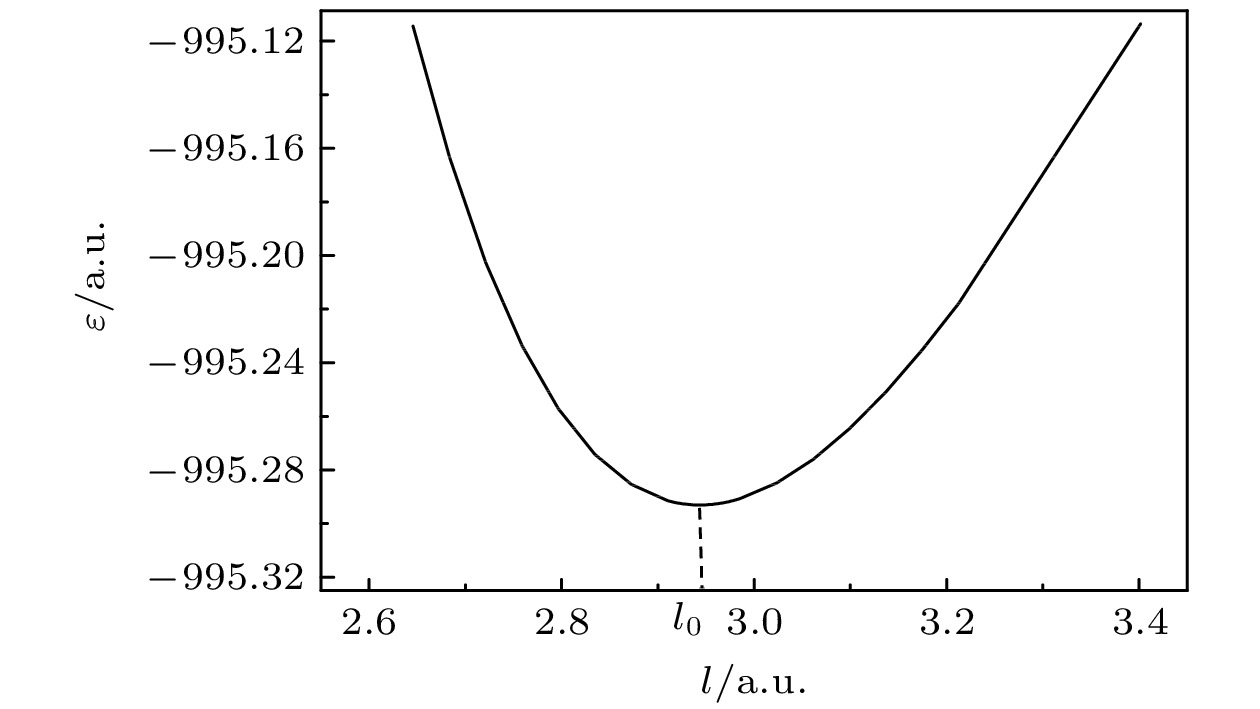
/6-311G*MP2/6-311G* CISD/6-311G* CCSD(T)/6-311G* B3LYP/6-311g* Expt[12] $ {\omega }_{1}/\mathrm{c}{\mathrm{m}}^{-1} $ 809 824 726 806 719 706 787 $ {\omega }_{2}/\mathrm{c}{\mathrm{m}}^{-1} $ 650 697 625 684 627 611 655 $ {\omega }_{3}/\mathrm{c}{\mathrm{m}}^{-1} $ 542 588 502 541 500 467 524 $ {\omega }_{4}/\mathrm{c}{\mathrm{m}}^{-1} $ 362 377 335 364 334 311 355 l0/(10–10m) 1.558 1.547 1.586 1.557 1.586 1.593 1.565 下载:
导出CSV
-
[1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] [21] [22]

 下载:
下载:
计量
- 文章访问数:6946
- PDF下载量:187
- 被引次数:0